8 (800) 333 02 98
ГОСТ 8269.1-97
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ЩЕБЕНЬ И ГРАВИЙ
ИЗ ПЛОТНЫХ ГОРНЫХ ПОРОД И ОТХОДОВ ПРОМЫШЛЕННОГО ПРОИЗВОДСТВА ДЛЯ СТРОИТЕЛЬНЫХ РАБОТ
Методы химического анализа
MAUNTAINOUS ROCK ROAD-METAL AND GRAVEL,
INDUSTRIAL WASTE PRODUCTS FOR CONSTRUCTION WORKS
Methods chemical analysis
Дата введения 1998-07-01
Предисловие
1 РАЗРАБОТАН институтом ВНИПИстромсырье с участием института НИИЖБ Российской Федерации
ВНЕСЕН Госстроем России
2 ПРИНЯТ Межгосударственной научно-технической комиссией по стандартизации, техническому нормированию и сертификации в строительстве (МНТКС) 10 декабря 1997 г.
За принятие проголосовали
Наименование государства | Наименование органа государственного управления строительством | ||||||||||
Азербайджанская Республика Республика Армения Республика Беларусь Республика Казахстан Кыргызская Республика Республика Молдова Российская Федерация Республика Таджикистан | Госстрой Азербайджанской Республики Министерство градостроительства Республики Армения Минстройархитектуры Республики Беларусь Агентство строительства и архитектурно-градостроительного контроля Министерства экономики и торговли Республики Казахстан Минархстрой Кыргызской Республики Министерство территориального развития, строительства и коммунального хозяйства Республики Молдова Госстрой России Госстрой Республики Таджикистан |
3 ВЗАМЕН ГОСТ 5578-94, ГОСТ 25592-91, ГОСТ 25818-91 в части методов химического анализа
4 ВВЕДЕН в действие с 1 июля 1998 г. в качестве государственного стандарта Российской Федерации постановлением Госстроя России от 6 января 1998 г. № 18-2
1 Область применения
Настоящий стандарт распространяется на щебень и гравий из плотных горных пород (в том числе попутно добываемых вскрышных и вмещающих пород и некондиционных отходов горных предприятий) и отходов промышленного производства (в том числе из топливных шлаков, шлаков черной и цветной металлургии, золошлаковых смесей и зол-уноса тепловых электростанций), применяемых в качестве заполнителей и компонентов тяжелых, легких и ячеистых бетонов, а также для дорожных и других видов строительных работ, и устанавливает методы определения массовой доли химических элементов и порядок выполнения химического анализа при оценке пригодности в строительстве отходов горных предприятий и промышленного производства.
2 Нормативные ссылки
Используемые в настоящем стандарте ссылки на нормативные документы приведены в приложении A.
3 Термины и определения
В настоящем стандарте применены следующие термины и определения.
Проба — определенное количество материала, отобранное для испытаний от партии горной породы, щебня (гравия) и отходов промышленного производства.
Аналитическая проба — проба материала, приготовленная из поступившей на испытание пробы и предназначенная для определения влаги и проведения полного химического анализа. Из аналитической пробы отбирают отдельные навески в соответствии с методикой испытаний. Допускается использование одной навески для проведения нескольких видов испытаний в соответствии с установленными методиками.
Навеска — проба материала, приготовленная из аналитической пробы и предназначенная для определения массовой доли элемента или нескольких элементов.
«Глухой» опыт — проведение испытания при отсутствии исследуемого материала с применением тех же реактивов и соблюдением всех условий анализа. На основании «глухого» опыта в результаты анализов вносят необходимые поправки.
4 Методы химического анализа
4.1 Общие положения
4.1.1 При проведении полного химического анализа щебня (гравия) определяют содержание кремния, алюминия, железа (трех- и двухвалентного), марганца, кальция, магния, калия, натрия, серы (сульфатной и сульфидной), хлоридов и потери массы при прокаливании (далее — п.п.п.).
В отдельных случаях (факультативно) определяют содержание титана, хрома, фосфора.
Анализируемую пробу переводят в раствор путем разложения, сплавлением или кислотами. Способ разложения пробы указан в конкретных методах настоящего стандарта.
Содержание кремния, алюминия, трехвалентного железа, кальция, магния определяют для одной навески после разложения ее сплавлением с карбонатом натрия или со смесью карбонатов натрия и калия.
Содержание марганца, хрома, титана и фосфора определяют фотоколориметрическим методом для другой навески после разложения ее сплавлением со смесью карбоната натрия и тетрабората натрия (буры).
Для отдельных навесок определяют п.п.п., калий и натрий пламенно-фотометрическим методом, двухвалентное железо, соединения серы, хлориды.
Содержание (массовую долю) всех компонентов, кроме хлоридов и сульфидов, представляют в виде соответствующих оксидов. Содержание сульфида представляют в пересчете на триоксид серы или серы S. Результаты анализа записывают в следующем порядке: п.п.п., SiO2, TiO2, Аl2О3, Fе2О3, FeO, Сr2О3, MnO, CaO, MgO, К2О, Na2O, Р2О5, SO3, S, Cl.
В случае, если элемент определяли, но он не был обнаружен, пишут «нет»; если какой-либо элемент не определяли, ставят прочерк.
Кроме выполнения полного химического анализа щебня (гравия), определяют сумму оксида и гидроксида кальция, называемую свободным оксидом кальция или свободной известью (CaOсвоб).
4.1.2 При проведении химического анализа щебня и гравия аналитическую пробу щебня и гравия из плотных горных пород отбирают в соответствии с разделом 4.2 ГОСТ 8269.0, отбор проб отходов промышленного производства осуществляют в соответствии с действующей нормативной или технической документацией на эти материалы.
Отобранную пробу сокращают до 100 г, измельчают до размера частиц, проходящих через сито № 016, и последовательным квартованием уменьшают до 50 г. Твердые зернистые материалы предварительно измельчают в металлической ступке до полного прохождения через сито № 05 по ГОСТ 6613, после чего магнитом удаляют попавшие в пробу металлические частицы.
Не допускается применение магнита для материалов, содержащих магнитные минералы. Дальнейшим квартованием отбирают для анализа среднюю аналитическую пробу массой свыше 10 до 25 г, которую растирают в агатовой, яшмовой или корундовой ступке до состояния пудры (при контрольном просеивании проба должна полностью проходить через сито № 008 по ГОСТ 6613).
Подготовленную пробу хранят в стеклянном бюксе с притертой крышкой для защиты от воздействия окружающей среды.
Перед взятием навески пробу высушивают в сушильном шкафу до постоянной массы при температуре (105±5) °С. Масса считается постоянной, если разность массы при двух последовательных взвешиваниях после сушки не превышает 0,0004 г.
В отдельных случаях, указанных в конкретных методиках настоящего стандарта, анализ проводят с навеской, находящейся в воздушно-сухом состоянии, с последующим пересчетом на сухое состояние. Массу навески т, г, находящейся в сухом состоянии, вычисляют по формуле
![]() , (1)
, (1)
где m0— масса навески материала в воздушно-сухом состоянии, г;
Х — массовая доля влаги в материале, определенная по 4.2.
4.1.3 Для взвешивания навесок в зависимости от допускаемой погрешности взвешивания применяют аналитические лабораторные весы общего назначения 2-го класса точности (типа ВЛР-200 или аналогичные) или 4-го класса точности (типа ВЛТК-500 или аналогичные) по ГОСТ 24104.
Массу навесок анализируемых проб, осадков в весовых методах, исходных веществ для приготовления стандартных растворов определяют с погрешностью не более 0,0002 г, массу навесок реактивов для приготовления титрованных растворов, индикаторов для приготовления растворов и индикаторных смесей — с погрешностью не более 0,001 г, массу навесок вспомогательных растворов — с погрешностью не более 0,01 г, массу плавней — с погрешностью не более 0,1 г.
4.1.4 Для проведения анализа применяют мерную лабораторную посуду не ниже 2-го класса точности по ГОСТ 29228, ГОСТ 29230 и ГОСТ 29252 (бюретки, пипетки) и ГОСТ 1770 (цилиндры, мензурки, колбы), а также стеклянную посуду (стаканы, колбы конические, воронки конические, эксикаторы и др.) по ГОСТ 25336, фарфоровую посуду и оборудование (тигли, лодочки, вставки для эксикаторов и др.) по ГОСТ 9147, тигли и чашки из платины по ГОСТ 6563, беззольные фильтры по соответствующей нормативной и технической документации.
Допускается применение аналогичной импортной посуды и материалов.
4.1.5 Для приготовления растворов и проведения анализов применяют реактивы не ниже класса ч.д.а., если не указана иная классификация, и дистиллированную воду, которая должна соответствовать ГОСТ 6709.
Горячая вода или горячий раствор, применяемые при химическом анализе, должны иметь температуру 60—80 °С, теплая вода или теплый раствор — 40—50 °С.
4.1.6 Для прокаливания и сплавления навесок анализируемых проб с плавнями применяют муфельные лабораторные электропечи или печи аналогичного типа с температурой нагрева (1000±50) °С.
Для сушки материалов используют сушильные шкафы с терморегулятором.
Для проведения анализов используют электрические плитки с закрытой спиралью, песчаные и водяные бани, термометры, магнитные мешалки, титраторы, фотоэлектротитриметры, иономеры, рН-метры, пламенные фотометры, концентрационные фотоэлектроколориметры, спектрофотометры.
Применяемые средства анализа должны соответствовать требованиям нормативной и технической документации на них.
4.1.7 Применяемые стандартизированные средства измерения должны быть поверены и аттестованы по ГОСТ 8.326, ГОСТ 8.513, ГОСТ 24555.
4.1.8 Концентрацию растворов выражают:
— в процентах по массе, численно равной массе растворенного вещества в граммах в 100 г раствора;
— массой растворенного вещества в граммах в 1 л раствора, г/л;
— массой растворенного вещества (или эквивалентного ему количества определяемого вещества) в граммах в 1 мл раствора, г/мл;
— в молях растворенного вещества в 1 л раствора (молярная концентрация М);
— в грамм-эквивалентах растворенного вещества в 1 л раствора (нормальная концентрация Н);
— соотношением объемных частей, например, 1:2, где первое число означает объемные части концентрированной кислоты или иного реактива, а второе — объемные части воды (если не указан другой растворитель).
Если в методике проведения анализа не указана концентрация или разбавление кислоты или водного раствора аммиака, то это концентрированная кислота или концентрированный раствор водного аммиака.
4.1.9 Массовую концентрацию стандартных растворов, а также титр титранта по определяемому веществу в г/мл и соотношение объемов растворов (в титриметрических методах) рассчитывают как среднеарифметическое по результатам не менее трех параллельных определений. Расчет проводят до четвертого знака после запятой.
4.1.10 Допускается последовательное определение нескольких элементов из одной навески, переведенной в раствор.
4.1.11 Для контроля погрешности результатов анализа используют изготовленные и аттестованные в соответствии с ГОСТ 8.315 и ГОСТ 8.532 стандартные образцы состава веществ и материалов: государственные и отраслевые стандартные образцы (ГСО и ОСО), стандартные образцы предприятия (СОП). При этом результат анализа стандартного образца считают удовлетворительным, если среднеарифметическое двух параллельных определений отличается от аттестованного значения массовой доли определяемого элемента не более чем на 0,7 ошибки повторяемости, установленной в стандарте для соответствующего элемента.
При отсутствии стандартных образцов контроль осуществляют по стандартным растворам.
4.1.12 Массовую долю элементов в анализируемой пробе определяют параллельно в двух навесках. За результат анализа принимают среднеарифметическое двух параллельных определений. При этом максимальное абсолютное расхождение между крайними результатами анализа не должно превышать абсолютных допустимых расхождений, установленных для конкретного метода.
4.1.13 В качестве норм точности (метрологических характеристик) определения содержания элемента используют расхождение между параллельными определениями.
Максимальные абсолютные расхождения результатов параллельных определений не должны превышать величину абсолютного допустимого расхождения для доверительной вероятности Р=0,95.
Средний результат анализа стандартного образца не должен отличаться от массовой доли определяемого элемента более чем на половину величины абсолютного допустимого расхождения для соответствующего интервала концентраций.
В случае, если соответствующими нормативными документами установлено предельное значение для определяемого элемента, а полученный результат анализа отличается от этого предельного значения более чем на величину допустимой ошибки повторяемости, следует произвести повторный анализ не менее трех навесок.
За окончательный результат принимают среднеарифметическое значение этих определений.
Если предельное значение установлено для суммы элементов (оксидов), то отличие полученного результата определения этой суммы от предельного значения оценивают по сумме допустимых ошибок повторяемости, установленных для элементов, умноженных на соответствующую долю элементов в полученной сумме.
4.1.14 Расхождение результатов определений в двух лабораториях не должно превышать допустимого расхождения между результатами двух параллельных определений.
Если расхождения между результатами параллельных испытаний пробы или стандартного образца, проводимых в лаборатории, превышают допускаемые величины, проводят повторные испытания.
Если при повторных испытаниях хотя бы одно из указанных расхождений превысит допустимую величину, результаты анализа признают неверными, испытания прекращают до выявления и устранения причин, вызвавших нарушения нормального хода анализа.
4.1.15 При применении физико-химических методов анализа, например фотоэлектроколориметрического, требующих построения градуировочных графиков, графики строят в прямоугольных координатах. На оси абсцисс откладывают величину массы определяемого элемента, г, мг, или массовую долю, %, на оси ординат — соответствующий аналитический сигнал (величину оптической плотности, силу тока и др.).
Для построения графиков используют ГСО, ОСО, состав которых близок к составу исследуемого материала, или реактивы, из которых готовят градуировочные растворы.
Условия построения графиков указаны в соответствующих методиках настоящего стандарта.
График строят не менее чем по пяти точкам, которые равномерно распределяют по диапазону измерений.
Минимальную и максимальную навески рассчитывают таким образом, чтобы обеспечить весь необходимый диапазон измерений. Каждую точку находят как среднеарифметическое значение не менее трех параллельных определений. Не допускается строить градуировочный график методом экстраполяции.
При использовании аликвотных частей массовую долю элемента (оксида) x0, %, определяют по формуле
![]() , (2)
, (2)
где т1 — масса элемента (оксида) в аликвотной части раствора, определенная по градуировочному графику, мг;
V — общий объем исходного раствора, мл;
т — масса навески, мг;
v1— аликвотная часть исходного раствора, мл.
4.1.16 При выполнении анализа навеску анализируемой пробы, разведение и аликвотные части необходимо принимать такими же, как при изготовлении основного градуировочного раствора.
В случае необходимости изменения навески, разведения или аликвотной части по сравнению с условиями приготовления основного градуировочного раствора вводят поправки, учитывающие эти изменения.
4.1.17 При фотоколориметрическом анализе вводят поправку на изменение условий фотометрирования по сравнению с условиями градуировки. Для этого одновременно с анализируемым образцом измеряют оптическую плотность вновь приготовленного окрашенного градуировочного раствора.
Измерение оптической плотности раствора выполняют с погрешностью не более ±0,002. Поправку вносят с обратным знаком:
если оптическая плотность градуировочного раствора увеличилась на несколько единиц, то это значение отнимают от величины оптической плотности анализируемого раствора, и наоборот. После введения поправки находят по графику искомую массовую долю элемента.
4.1.18 Проверка градуировочных графиков должна проводиться периодически по стандартным образцам по соответствующим методикам настоящего стандарта не реже одного раза в полугодие, а также после каждого ремонта используемых приборов.
4.1.19 В фотоколориметрических методах содержание определяемого оксида допускается находить методом сравнения оптической плотности исследуемого раствора (или его аликвотной части) с оптической плотностью стандартного раствора близкой концентрации.
Концентрация колориметрируемого раствора не должна отличаться от концентрации стандартного более чем в 1,2 раза.
В случае нарушения этого условия меняют навеску, разведение или аликвотную часть анализируемой пробы или стандартного образца раствора.
Массовую долю оксида x0, %, определяют по формуле
![]() , (3)
, (3)
где V — объем окрашенной фотометрируемой части исследуемого раствора, мл;
V0 — общий объем исследуемого раствора, мл;
т — масса навески, г;
Vа — объем аликвотной части исследуемого раствора, взятого для приготовления окрашенного раствора при фотометрировании, мл;
Сх — концентрация окрашенной фотометрируемой части исследуемого раствора, мг/мл, определяемая по формуле
![]() , (4)
, (4)
где Сст — концентрация стандартного раствора, мг/мл;
Дх и Дст— оптические плотности соответственно исследуемого и стандартного растворов.
4.1.20 При выполнении анализов рекомендуется параллельно проводить «глухой» опыт для учета загрязнений реактивов, дистиллированной воды и др.
4.1.21 Содержание вредных веществ в воздухе рабочей зоны, лаборатории, образующихся в ходе анализа, не должно превышать предельно допустимых концентраций, указанных в ГОСТ 12.1.005.
4.1.22 Контроль за содержанием вредных веществ в воздухе рабочей зоны лаборатории проводят по ГОСТ 12.1.005 и ГОСТ 12.1.007.
4.1.23 При работе с горючими и взрывоопасными веществами должны соблюдаться требования безопасности в соответствии с ГОСТ 12.1.010.
4.1.24 Все используемые электрические приборы должны соответствовать правилам устройства электроустановок (ПУЭ).
Эксплуатацию электрических приборов проводят в соответствии с правилами техники безопасности при эксплуатации электроустановок, а также правилами электробезопасности по ГОСТ 12.1.019.
4.1.25 Пожарная безопасность лабораторных помещений должна обеспечиваться в соответствии с ГОСТ 12.1.004, безопасность труда — в соответствии с ГОСТ 12.1.007.
4.1.26 При использовании в качестве реактивов опасных (едких, токсичных) веществ следует руководствоваться требованиями безопасности, изложенными в нормативных или технических документах на эти реактивы, применять индивидуальные средства защиты (респираторы) по ГОСТ 12.4.011 или ГОСТ 12.4.028, резиновые перчатки по ГОСТ 12.4.103, одежду по ГОСТ 27654 и ГОСТ 29058.
4.2 Определение влаги
Содержание влаги определяют весовым методом по разности между массой бюксы с навеской до и после высушивания.
4.2.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью измерения ±0,0002 г.
Эксикатор по ГОСТ 25336.
Шкаф сушильный.
Бюксы по ГОСТ 23932.
Кальций хлористый (хлорид кальция) по ГОСТ 450, прокаленный при температуре 700—800 °С, для заполнения эксикатора.
4.2.2 Порядок проведения испытания
Навеску массой 1 г помещают в предварительно высушенную до постоянной массы бюксу, ставят в сушильный шкаф, нагретый до температуры (105±5) °С, сушат 1,5—2 ч, после чего охлаждают в эксикаторе и взвешивают.
Перед взвешиванием крышку бюксы приоткрывают и быстро закрывают. Высушивание и охлаждение повторяют до тех пор, пока разность массы между двумя последующими взвешиваниями будет не более 0,0004 г.
Если при повторном высушивании масса навески увеличится, для расчета применяют массу, предшествующую ее увеличению.
4.2.3 Обработка результатов испытания
Массовую долю влаги X, %, определяют по формуле
![]() , (5)
, (5)
где m1 — масса бюксы с навеской до сушки, г;
т2 — масса бюксы с навеской после сушки, г;
m0— масса навески, г.
Абсолютное допустимое расхождение между результатами двух параллельных определений не должно превышать, %:
— 0,10 при содержании влаги до 1,0 % по массе;
— 0,20 » » » св. 1,0 » » »
4.3 Определение потери массы при прокаливании
Потерю массы при прокаливании определяют весовым методом по разности массы тигля с навеской исследуемой пробы щебня (гравия) до и после прокаливания.
4.3.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью взвешивания ±0,0002 г.
Печь муфельная с номинальной температурой (1000±50) °С.
Эксикатор по ГОСТ 25336.
Фарфоровый тигель по ГОСТ 9147.
4.3.2 Порядок проведения испытания
Пробу подготавливают в соответствии с 4.1.2. Из подготовленной пробы, находящейся в сухом состоянии, отбирают навеску массой 1 г, которую помещают в предварительно прокаленный до постоянной массы фарфоровый тигель и взвешивают. Затем навеску помещают в муфельную печь и прокаливают в течение 2 ч при температуре (1000±50) °С.
После прокаливания тигель охлаждают в эксикаторе и взвешивают. Прокаливание повторяют до достижения постоянной массы. Если при повторном прокаливании масса навески увеличивается, для расчета принимают величину массы до ее увеличения.
4.3.3 Обработка результатов анализа
Потерю массы при прокаливании (п.п.п.), %, определяют по формуле
![]() , (6)
, (6)
где т1 — масса исходной навески в сухом состоянии, вычисленная по разности масс тигля с пробой и без нее до прокаливания, г;
т2 — масса прокаленного остатка, вычисленная по разности масс тигля с пробой и без нее по окончании прокаливания, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 1.
В случае, когда масса навески увеличивается после первого прокаливания, что возможно при наличии двухвалентного железа, марганца и других элементов низких степеней окисления, потери при прокаливании определяют по разности между 100 % (принята масса навески) и суммой всех определенных элементов.
Таблица 1 В процентах
Потеря массы при прокаливании | Абсолютное допустимое расхождение | ||||||||||
До 1,0 включ. От 1,0 » 10,0 Св. 10,0 | 0,10 0,20 0,30 |
4.4 Определение диоксида кремния
Метод основан на разложении анализируемой пробы сплавлением и определении диоксида кремния весовым методом с обязательным последующим удалением его в виде фторида кремния.
4.4.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью взвешивания ±0,0002 г.
Печь муфельная.
Тигли платиновые по ГОСТ 6563.
Эксикатор по ГОСТ 25336.
Стаканы вместимостью 150—200 мл по ГОСТ 25336.
Воронки по ГОСТ 25336.
Натрий углекислый (карбонат натрия) безводный по ГОСТ 83.
Калий углекислый (карбонат калия) по ГОСТ 4221.
Кислота соляная по ГОСТ 3118 плотностью 1,19, раствор 5:95.
Кислота серная по ГОСТ 4204 плотностью 1,84.
Кислота фтористоводородная (плавиковая) по ГОСТ 10484, 40 %-ная.
Желатин пищевой, 1 %-ный раствор.
Серебро азотнокислое (нитрат серебра) по ГОСТ 1277, 1 %-ный раствор, подкисленный 2—3 каплями азотной концентрированной кислоты на 100 мл раствора.
Кислота азотная концентрированная по ГОСТ 4461.
Аммоний углекислый (карбонат аммония) по ГОСТ 3762.
Фильтры «белая лента».
Плавень — натрий углекислый (карбонат натрия) или смесь равных количеств по массе карбонатов натрия и калия.
4.4.2 Порядок проведения анализа
Навеску массой 0,3 г помещают в платиновый тигель, перемешивают с плавнем, взятым в «шестикратном (по массе) количестве, накрывают крышкой и ставят в муфельную печь.
При применении в качестве плавня углекислого натрия навеску сплавляют при температуре 1000 °С, смеси щелочных карбонатов — при 800 °С.
При высоком содержании в пробе двухвалентного железа для обеспечения полного перехода его в трехвалентную форму в плавень можно добавить нитрат аммония в количестве 1 % массы плавня. Плав выдерживают в муфельной печи 15 мин. После этого тигель опускают в холодную воду так, чтобы в него не попала вода.
Охлажденный плав извлекают из тигля следующим образом. В тигель наливают около 7—10 мл горячей воды, накрывают крышкой и выдерживают. Если в плаве образуется королек, его переносят в стакан вместимостью 150—200 мл. Если королек не образовался, плав извлекают постепенно, добавляя в тигель маленькими порциями (по несколько капель) соляную кислоту, помешивая палочкой.
Чтобы кислота не разбрызгивалась, тигель следует прикрывать крышкой. На все извлечение плава расходуется 25—30 мл соляной кислоты.
После того, как весь плав будет перенесен, тигель обмывают кислотой и обтирают кусочками фильтра. В стакан приливают 5 мл раствора желатина и в течение 5 мин хорошо перемешивают.
Тигель с крышкой, стекло и стенки стакана обмывают горячей водой (30—50 мл). Стакан накрывают стеклом и ставят в теплое место на 25—30 мин для коагуляции осадка.
Когда раствор над осадком станет прозрачным, его отфильтровывают через неплотный фильтр. Осадок промывают 2—3 раза горячим раствором соляной кислоты (5:95) декантацией, а затем на фильтре горячей водой до исчезновения реакции на ион хлора.
Несколько капель фильтрата помещают на часовое стекло. Если при добавлении капли раствора азотнокислого серебра (нитрата серебра) образуется взвесь, то проба не отмыта.
Как только реакция на ион хлора станет отрицательной, фильтрат выпаривают для вторичного осаждения диоксида кремния. Фильтрат выпаривают досуха, затем приливают 20 мл соляной кислоты, добавляют 5 мл раствора желатина и перемешивают в течение 5 мин. После этого стенки стакана обмывают горячей водой и ставят его на 30 мин в теплое место для коагуляции осадка.
Затем осадок отфильтровывают через неплотный фильтр, как при первом осаждении. Осадки от первого и второго осаждения соединяют и помещают во взвешенный платиновый тигель. Осторожно озоляют и прокаливают в муфельной печи при температуре 1000— 1100 °С в течение 45—60 мин до получения постоянной массы, охлаждают в эксикаторе и взвешивают.
Прокаленный осадок смачивают несколькими каплями воды, прибавляют 1—2 мл серной и 5—7 мл плавиковой кислоты и выпаривают на плитке несильного накала (чтобы кислота не разбрызгивалась) до прекращения выделения паров серной кислоты. После этого тигель прокаливают при температуре 1000—1100 °С в течение 15 мин, охлаждают в эксикаторе и взвешивают.
Если масса осадка более 0,01 мг, его сплавляют и присоединяют к фильтрату, который переносят в мерную колбу вместимостью 250 мл, доводят водой до метки и в дальнейшем используют для определения оксидов железа, алюминия, кальция и магния.
4.4.3 Обработка результатов анализа
Массовую долю диоксида кремния SiO2 %, определяют по формуле
![]() , (7)
, (7)
где т1 — масса осадка до отгонки плавиковой кислоты, г;
т2— масса осадка после отгонки плавиковой кислоты, г;
т — масса сухой навески, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 2.
Таблица 2 В процентах
Массовая доля диоксида кремния | Абсолютное допустимое расхождение | ||||||||||
От 1,0 до 5,0 включ. | 0,15 | ||||||||||
Св. 5,0 » 18,0 | 0,25 | ||||||||||
» 18,0 » 25,0 | 0,30 | ||||||||||
» 25,0 » 40,0 | 0,40 | ||||||||||
» 40,0 » 70,0 | 0,50 | ||||||||||
» 70,0 | 0,60 |
4.5 Определение оксидов железа и алюминия
Метод основан на комплексометрическом определении оксидов железа и алюминия после предварительного отделения диоксида кремния по 4.5.1 или непосредственно из отдельной навески по 4.5.2.
Сущность метода заключается в способности комплексона III (динатриевой соли этилендиамин — N, N, N1, N1 — тетрауксусной кислоты — трилона Б) образовывать комплексы с ионами Fe3+ и Аl3+. Комплексонат железа (III) возникает при рН=1—1,5.
В качестве индикатора применяют сульфосалициловую кислоту, которая в сильнокислой среде дает с ионами трехвалентного железа растворимый сульфосалицилат железа, окрашенный в фиолетовый цвет.
В точке эквивалентности окраска сульфосалицилата железа исчезает.
Содержание оксида алюминия находят в той же пробе обратным титрованием. Для этого после определения железа в раствор приливают трилон Б в количестве, большем, чем надо для связывания алюминия в комплекс, а избыток трилона Б оттитровывают хлоридом железа (III).
4.5.1 Определение оксидов железа и алюминия после предварительного отделения диоксида кремния
4.5.1.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью взвешивания ±0,0002 г.
Печь муфельная.
Колбы мерные вместимостью 1 л по ГОСТ 1770.
Бюретки по ГОСТ 29251 и ГОСТ 29252.
Пипетки по ГОСТ 29227 или ГОСТ 29228.
Колбы конические вместимостью 250 мл по ГОСТ 25336.
Стаканы вместимостью 150 мл по ГОСТ 25336.
Воронки по ГОСТ 25336.
Электроплитка.
Бутыль полиэтиленовая вместимостью 10 л.
Тигли фарфоровые по ГОСТ 9147.
Кислота соляная по ГОСТ 3118 плотностью 1,19, раствор 1:3.
Аммиак водный по ГОСТ 3760, 10- и 25 %-ные водные растворы.
Железо (III) хлорид 6-водный по ГОСТ 4147.
Кислота сульфосалициловая 2-водная по ГОСТ 4478.
Алюминий марки A995 поГОСТ 11069, стружка.
Кислота уксусная по ГОСТ 18270.
Натрий уксуснокислый 3-водный (ацетат натрия) по ГОСТ 199.
Соль динатриевая этилендиамин — N, N, N1, N1 — тетрауксусной кислоты 2-водная (трилон Б) по ГОСТ 10652, раствор 0,025 М.
Индикаторная бумага Конго.
Аммоний азотнокислый (нитрат аммония) по ГОСТ 22867, 2 %ный раствор.
Индикатор метиловый красный, 2 %-ный спиртовой раствор.
Титрованный раствор соли трехвалентного железа:
6,76 г хлорида железа (III) FеСl3(6Н2О растворяют в 300 мл воды. Раствор фильтруют в мерную колбу вместимостью 1 л, добавляют 15 мл соляной кислоты плотностью 1,19 и доливают водой до метки.
Титр раствора хлорида железа (III) устанавливают весовым методом. Для этого отбирают пипеткой 50 мл раствора хлорида железа (III), переносят его в стакан, добавляют 2—3 капли индикатора метилового красного, ставят его на плитку и нагревают до кипения. Затем, сняв стакан с плитки, осаждают гидроксид железа раствором аммиака.
Раствор аммиака добавляют в таком количестве, которое обеспечит изменение окраски раствора из розовой в желтую и появление слабого запаха аммиака. Стакан выдерживают в теплом месте для коагуляции осадка 10 мин, после чего отфильтровывают осадок через неплотный фильтр «красная лента». Осадок на фильтре промывают 8—10 раз горячим раствором азотнокислого аммония (нитратом аммония). Затем осадок вместе с фильтром переносят в тигель, подсушивают и прокаливают в муфельной печи при температуре 1000 °С в течение 20—25 мин до постоянной массы.
Титр раствора хлорида железа (III) по Fe2О3, г/мл, определяют по формуле
![]() , (8)
, (8)
где т — масса прокаленного осадка, г.
Сульфосалициловая кислота, 20 %-ный раствор:
20 г кислоты растворяют в 50 мл воды, нейтрализуют 25 %-ным раствором аммиака до изменения окраски индикаторной бумаги Конго из синего в фиолетовый цвет и разбавляют водой до 100 мл.
Точный раствор соли алюминия:
0,6745 г чистого металлического алюминия растворяют в 11,2 мл 25 %-ного раствора соляной кислоты и доводят водой до 1 л. Титр полученного раствора по Al2O3 равен 0,0012745 г/мл.
Буферный раствор:
270 г уксуснокислого натрия (ацетата натрия) растворяют в 300 мл воды, фильтруют, разбавляют водой до 500 мл, добавляют 500 мл раствора уксусной кислоты, содержащего 70 мл концентрированной или 90 мл 30 %-ной уксусной кислоты, и тщательно перемешивают.
Титрованный 0,025 М раствор трилона Б:
95 г трилона Б растворяют в 1 л воды, фильтруют в полиэтиленовую бутыль, разбавляют водой до 10 л и тщательно перемешивают.
Титр раствора трилона Б по Fe2O3 устанавливают следующим образом: из бюретки наливают 20 мл раствора хлорида железа (III) в коническую колбу вместимостью 250 мл, разбавляют его водой до 100 мл, нагревают до 50—70 °С, добавляют 7—8 капель сульфосалицилового индикатора и титруют раствором трилона Б до исчезновения фиолетового цвета сульфосалицилата железа.
Титр раствора трилона Б по Fе2О3, г/мл, определяют по формуле
![]() , (9)
, (9)
где ![]() — титр раствора хлорида железа (III), г/мл;
— титр раствора хлорида железа (III), г/мл;
V — объем раствора трилона Б, идущего на титрование, мл.
Перед определением титра раствора трилона Б по оксиду алюминия находят соотношение между концентрациями раствора трилона Б и хлорида железа (III). Для этого в три конические колбы вместимостью 250 мл наливают из бюретки по 10 мл раствора трилона Б, разбавляют его водой до 100 мл, добавляют 10 мл буферного раствора, 7—8 капель сульфосалициловой кислоты и титруют раствором хлорида железа (III) до появления золотисто-оранжевого цвета, не исчезающего в течение 1 мин.
По среднему результату трех титрований вычисляют коэффициент соотношения К между концентрациями растворов хлорида железа (III) и трилона Б:
![]() , (10)
, (10)
где V — объем раствора хлорида железа (III), идущего на титрование 10 мл раствора трилона Б, мл.
Далее определяют титр раствора трилона Б по Al2O3.
В три конические колбы вместимостью 250 мл наливают из бюретки по 25 мл точного раствора соли алюминия, разбавляют его водой до 100 мл, нейтрализуют 10 %-ным раствором аммиака до перехода окраски бумаги Конго в красный цвет. Затем добавляют по капле соляную кислоту (1:3), пока не изменится окраска бумаги Конго в синий цвет, после чего добавляют 8—10 капель соляной кислоты.
К полученному раствору доливают 30 мл раствора трилона Б, нагревают до кипения, прибавляют 10 мл буферного раствора, 7—8 капель сульфосалициловой кислоты. Затем охлаждают до комнатной температуры и титруют раствором хлорида железа (III) до появления золотисто-оранжевого цвета, не исчезающего в течение 1 мин.
Титр раствора трилона Б по Аl2O3, г/мл, определяют по формуле
![]() , (11)
, (11)
где Т — титр раствора соли алюминия по Аl2O3, г/мл;
К — коэффициент соотношения между концентрациями растворов трилона Б и хлорида железа;
V — объем раствора хлорида железа (III), идущего на титрование, мл.
4.5.1.2 Порядок проведения анализа
Для определения железа и алюминия отбирают пипеткой точно 50—100 мл фильтрата от диоксида кремния (4.4.2) и помещают его в коническую колбу вместимостью 250 мл, добавляют 2—3 капли азотной кислоты, нагревают до температуры 50—60 °С, опускают в него кусочек индикаторной бумаги Конго и нейтрализуют раствором аммиака до изменения окраски бумаги из синего цвета в красный. Затем добавляют по капле раствор соляной кислоты (1:3), пока не изменится окраска бумаги Конго из красного в фиолетовый цвет, добавляют еще 15 капель соляной кислоты, 4—6 капель раствора сульфосалициловой кислоты и титруют раствором трилона Б до обесцвечивания раствора. В эквивалентной точке раствор становится или бесцветным, или светло-желтым, при этом раствор не должен иметь розового оттенка.
Оттитровав железо, добавляют из бюретки трилон Б в таком количестве, чтобы избыток его после образования комплекса с алюминием был 10 мл или немного больше.
Раствор нагревают до кипения, а затем охлаждают до 60 °С, нейтрализуют буферным раствором до изменения синей окраски бумаги Конго в красный цвет и еще вводят 10 мл буферного раствора. Раствор охлаждают до комнатной температуры и титруют раствором хлорида железа (III) до появления устойчивой бурой окраски, не исчезающей в течение 1—1,5 мин.
4.5.1.3 Обработка результатов анализа
Массовую долю оксида железа (III) Fе2О3, %, когда в анализируемой пробе отсутствует оксид железа (II), определяют по формуле
![]() , (12)
, (12)
где V — объем раствора трилона Б, пошедшего на титрование, мл;
![]() — титр раствора трилона Б по Fe2O3, г/мл;
— титр раствора трилона Б по Fe2O3, г/мл;
т — масса навески, г.
При анализе материалов, содержащих двух- и трехвалентное железо, массовую долю оксида трехвалентного железа Fе2О3 (III), %, определяют по формуле
Fе2О3 (III) = Fе2О3 ( 1,1114 ( FеО, (13)
где Fе2О3 — содержание общего железа в пересчете на оксид железа (III), определенное по формуле (12), %;
FeO — содержание оксида железа (II), определяемое по формуле (28) %;
1,1114 — коэффициент пересчета содержания оксида железа (II) на оксид железа (III).
Массовую долю оксида алюминия Al2O3, %, определяют по формуле
![]() , (14)
, (14)
где v0 — объем раствора трилона Б, добавленного после определения оксида железа, мл;
V1— объем раствора хлорида железа (III), идущего на обратное титрование, мл;
К — коэффициент соотношения между концентрациями растворов трилона Б и хлорида железа (III);
![]() — титр раствора трилона Б по Al2O3, г/мл.
— титр раствора трилона Б по Al2O3, г/мл.
Абсолютное допустимое расхождение результатов параллельных определений содержания оксида железа и оксида алюминия не должно превышать значений, указанных в таблице 3.
Таблица 3 В процентах
Массовая доля | Абсолютное допустимое расхождение | ||||||||||
Оксид железа: | |||||||||||
до 1,0 включ. | 0,10 | ||||||||||
св. 1,0 » 3,0 » | 0,15 | ||||||||||
» 3,0 » 7,0 » | 0,20 | ||||||||||
» 7,0 » 25,0 » | 0,30 | ||||||||||
» 25,0 | 0,80 | ||||||||||
Оксид алюминия: | |||||||||||
от 1,0 до 3,0 включ. | 0,15 | ||||||||||
св. 3,0 » 7,0 » | 0,20 | ||||||||||
» 7,0 » 20,0 » | 0,30 | ||||||||||
» 20,0 » 70,0 » | 0,40 | ||||||||||
» 70,0 | 0,50 |
4.5.2 Определение оксидов железа и алюминия из отдельной навески комплексометрическим методом
4.5.2.1 Средства контроля и вспомогательное оборудование
Средства контроля и вспомогательное оборудование по 4.5.1.1 со следующими дополнениями:
тигель платиновый по ГОСТ 6563;
натрий углекислый (карбонат натрия) безводный по ГОСТ 83.
4.5.2.2 Порядок проведения анализа
Навеску массой 0,1—0,2 г помещают в платиновый тигель и спекают в муфельной печи при температуре 1000 °С в течение 5 мин с таким же количеством безводного карбоната натрия.
Спек разминают в тигле палочкой, смачивают 5 мл концентрированной соляной кислоты, выдерживают 15 мин в теплом месте и выщелачивают в стакан вместимостью 150—200 мл соляной кислотой (1:3). Раствор разбавляют водой до 100—150 мл и определяют железо и алюминий, как указано в 4.5.1.3.
4.5.2.3 Обработка результатов анализа
Обработка результатов анализа — по 4.5.1.3.
Абсолютное допустимое расхождение результатов параллельных определений содержания оксидов железа и алюминия не должно превышать значений, указанных в таблице 3.
4.6 Определение оксидов кальция и магния
Содержание оксидов кальция и магния определяют в фильтрате от диоксида кремния. Метод основан на способности катионов кальция и магния образовывать с трилоном Б при определенных значениях рН прочные комплексные соединения. При комплексометрическом титровании используют металлиндикаторы.
В момент достижения эквивалентной точки, когда весь катион связывается трилоном Б, появляется окраска свободного индикатора, отличающаяся от окраски комплексного с ним соединения.
Для чистоты анализа элементы группы полуторных оксидов отделяют осаждением уротропином по 4.6.1.
4.6.1 Отделение элементов группы полуторных оксидов уротропином
4.6.1.1 Средства контроля и вспомогательное оборудование
Электроплитка с закрытой спиралью.
Пипетка по ГОСТ 29227 или по ГОСТ 29228.
Стаканы вместимостью 150—200 мл по ГОСТ 25336.
Воронки по ГОСТ 25336.
Фильтры «белая лента».
Кислота соляная по ГОСТ 3118 плотностью 1,19, раствор 1:3.
Кислота азотная по ГОСТ 4461 плотностью 1,4.
Аммиак водный по ГОСТ 3760, 10 %-ный водный раствор.
Уротропин фармакопейный, 0,5- и 10 %-ный водные растворы.
Индикаторная бумага Конго.
Металлиндикатор кислотный хром темно-синий.
4.6.1.2 Порядок проведения анализа
Отбирают пипеткой 50 мл фильтрата от диоксида кремния, полученного по 4.4.2 настоящего стандарта, и помещают в стакан.
В стакан добавляют 3—5 капель азотной кислоты, бросают кусочек бумаги Конго и нейтрализуют аммиаком до начала ее покраснения.
После этого добавляют несколько капель соляной кислоты (1:3) до посинения бумаги Конго (можно нейтрализовать до появления взвеси и затем кислотой ее растворить).
Приливают 20 мл 10 %-ного раствора уротропина и нагревают в течение 10 мин при температуре 80—90 °С, не доводя раствор до кипения. Как только осадок коагулируется, его отфильтровывают через фильтр «белая лента» и промывают теплым 0,5 %-ным раствором уротропина.
4.6.2 Комплексометрическое титрование оксидов кальция и магния с металлиндикатором кислотным хромом темно-синим
Метод основан на комплексометрическом титровании ионов кальция и магния раствором трилона Б с металлиндикатором кислотным хромом темно-синим в одной пробе с одним и тем же индикатором, но при разных значениях рН раствора.
4.6.2.1 Средства контроля и вспомогательное оборудование
Колбы мерные вместимостью 100 мл и 1 л по ГОСТ 1770.
Пипетки по ГОСТ 29227 или по ГОСТ 29228.
Бюретки по ГОСТ 29251 или по ГОСТ 29252.
Колбы конические вместимостью 200—250 мл по ГОСТ 25336.
Натрия гидроокись (гидроксид натрия) по ГОСТ 4328, 20 %-ный раствор.
Сахароза по ГОСТ 5833, 2 %-ный раствор.
Кислота соляная по ГОСТ 3118, растворы 1:3, 1:5.
Аммоний хлористый (хлорид аммония) по ГОСТ 3773.
Аммиак водный по ГОСТ 3760, 25 %-ный раствор.
Кальций углекислый (карбонат кальция) по ГОСТ 4530 или стандартный образец известняка.
Магний сернокислый 7-водный (сульфат магния) по ГОСТ 4523 или стандарт-титр.
Спирт этиловый по ГОСТ 18300.
Индикаторная бумага Конго.
Аммиачный буферный раствор:
растворяют 70 г хлорида аммония в 200 мл воды, фильтруют, добавляют 570 мл 25 %-ного раствора аммиака, затем добавляют воды до 1 л и тщательно перемешивают (рН раствора равно 10).
Кислотный хром темно-синий металлиндикатор:
растворяют 0,5 г кислотного хрома темно-синего в 10 мл аммиачного буферного раствора и добавляют этилового спирта до 100 мл.
Трилон Б по ГОСТ 10652, 0,025 М раствор.
Титр раствора трилона Б по оксиду кальция устанавливают по химически чистому карбонату кальция или стандартному образцу известняка. Отвешивают 0,1 г высушенного карбоната кальция или стандартного образца известняка и вносят в коническую колбу вместимостью 250 мл, приливают 50 мл воды и осторожно небольшими дозами добавляют 15—20 мл соляной кислоты (1:5). Раствор нейтрализуют 20 %ным раствором гидроксида натрия по индикаторной бумаге Конго до слабощелочной среды, добавляют 10 мл избытка щелочи. Затем вносят 10 капель индикатора хрома темно-синего и титруют раствором трилона Б до изменения розовой окраски в сиренево-синюю.
Титр раствора трилона Б по СаО, г/мл, определяют по формуле
![]() , (15)
, (15)
где т — масса сухой навески карбоната кальция или стандартного образца, г;
С — содержание СаО в карбонате кальция или в стандартном образце, %;
V — объем раствора трилона Б, идущего на титрование, мл.
За результат принимают среднеарифметическое трех титрований.
Титр раствора трилона Б по оксиду магния устанавливают по химически чистому сульфату магния MgSО4 ( 7H2O или по стандарт-титру MgSО4 ( 7H2O. Отвешивают 0,1 г сульфата магния и вносят в коническую колбу вместимостью 250 мл, добавляют 50 мл воды, 10 мл аммиачного буферного раствора, 10 капель индикатора кислотного хрома темно-синего и титруют раствором трилона Б до изменения розовой окраски в сине-голубую.
Титр раствора трилона Б по MgO, г/мл, определяют по формуле
![]() , (16)
, (16)
где т — масса навески MgSО4 ( 7H2O, г;
С — содержание оксида магния в MgSО4 ( 7H2O, %;
V1 — объем раствора трилона Б, идущего на титрование магния, мл.
За результат принимают среднеарифметическое трех титрований.
4.6.2.2 Порядок проведения анализа
К фильтрату, полученному по 4.6.1.2, после отделения элементов группы полуторных оксидов уротропином прибавляют 10 мл 2 %-ного раствора сахарозы, если содержание СаО в пробе больше 10 %, или 5 мл, если содержание СаО в пробе не превышает 10 %. Раствор осторожно нейтрализуют 20 %-ным раствором гидроксида натрия до покраснения бумаги Конго и еще добавляют 10 мл раствора гидроксида натрия.
Затем прибавляют воды до общего объема приблизительно 100 мл и после тщательного перемешивания раствор выдерживают 1—2 мин для формирования осадка гидроксида магния. Затем прибавляют 10 капель раствора индикатора кислотного хрома темно-синего и, сильно перемешивая, медленно титруют раствором трилона Б до перехода окраски из розовой в неизменяющийся сиренево-синий цвет. Для определения магния после титрования кальция добавляют в испытываемый раствор 5 мл соляной кислоты (1:3), чтобы полностью растворился гидроксид магния, хорошо перемешивают и .смывают стенки колбы небольшим количеством воды. Раствор при этом меняет цвет на розовый. Бумага Конго должна оставаться красной. Если она посинеет, следует добавить по капле 20 %-ный раствор гидроксида натрия, пока она снова не покраснеет. Затем вводят 10 мл аммиачного буферного раствора и продолжают титрование трилоном Б до перехода цвета раствора из розового в устойчивый сине-голубой.
4.6.2.3 Обработка результатов анализа
Массовую долю оксида кальция СаО, %, определяют по формуле
![]() , (17)
, (17)
где TCaO — титр раствора трилона Б по СаО, г/мл ;
V — объем раствора трилона Б, израсходованного на титрование оксида кальция, мл ;
т — масса навески, г.
Массовую долю оксида магния MgO, %, рассчитывают по формуле
![]() , (18)
, (18)
где Т MgO — титр раствора трилона Б по MgO, г/мл;
V1— объем раствора трилона Б, израсходованного на титрование MgO, мл;
m — масса навески, г.
Абсолютное допустимое расхождение результатов параллельных определений содержания оксида кальция и оксида магния не должно превышать значений, указанных в таблице 4.
Таблица 4 В процентах
Массовая доля | Абсолютное допустимое расхождение | ||||||||||
Оксид кальция: | |||||||||||
до 1,0 включ. | 0,10 | ||||||||||
св. 1,0 » 5,0 » | 0,15 | ||||||||||
» 5,0 » 10,0 » | 0,20 | ||||||||||
» 10,0 » 40,0 » | 0,30 | ||||||||||
» 40,0 » 80,0 | 0,40 | ||||||||||
Оксид магния: | |||||||||||
до 1,0 включ. | 0,15 | ||||||||||
св. 1,0 » 6,0 » | 0,30 | ||||||||||
» 6,0 » 25,0 | 0,60 |
4.6.3 Определение оксидов кальция и магния при наличии в щебне (гравии) соединений марганца
При наличии в пробе оксида марганца свыше 0,2 до 2,0 % по массе содержание кальция определяют комплексометрическим титрованием с индикатором мурексидом в присутствии восстановителя — солянокислого гидроксиламина.
Метод не применяется для определения содержания кальция и магния при наличии в исследуемом материале (пробе) марганца свыше 2,0 % по массе, который необходимо предварительно отделить.
При наличии в пробе оксида марганца до 0,2 % по массе содержание оксидов кальция и магния определяют по 4.6.2.
4.6.3.1 Средства контроля и вспомогательное оборудование.
Колбы, пипетки, бюретки по 4.6.2.1.
Натрия гидроокись (гидроксид натрия) по ГОСТ 4328, 20 %-ный раствор.
Гидроксиламина гидрохлорид по ГОСТ 5456, 5 %-ный водный раствор.
Натрий хлористый (хлорид натрия) по ГОСТ 4233.
Кислота соляная по ГОСТ 3118, 10%-ный раствор.
Мурексид, сухая смесь: 1 г мурексида смешивают с 99 г натрия хлористого (хлорида натрия).
Трилон Б — соль динатриевая этилендиамин — N, N, N1, N1 — тетрауксусной кислоты 2-водная по ГОСТ 10652 — 0,05 М раствор. Титр раствора трилона Б по MgO устанавливают по 4.6.2.1, титр раствора трилона Б по СаО устанавливают по 4.6.2.1, при этом вместо индикатора кислотного хрома темно-синего применяют индикатор мурексид.
4.6.3.2 Порядок проведения анализа
К фильтрату после отделения элементов группы полуторных оксидов по 4.6.1 прибавляют 1—2 мл раствора гидроксиламина гидрохлорида, перемешивают, добавляют дистиллированной воды до 100 мл, 10—15 мл раствора гидроксида натрия, снова перемешивают и вносят на кончике шпателя мурексид. Затем медленно титруют раствором трилона Б до перехода малиновой окраски в фиолетовую.
После определения кальция в той же пробе определяют магний, предварительно изменив окраску мурексида путем введения соляной кислоты. Магний титруют трилоном Б с индикатором кислотным хромом темно-синим, как указано в 4.6.2.2.
4.6.3.3 Обработка результатов анализа
Обработку результатов проводят по 4.6.2.3.
Абсолютное допустимое расхождение результатов параллельных определений содержания оксида кальция и оксида магния не должно превышать значений, указанных в таблице 4.
4.7 Определение сульфатной и сульфидной серы
Содержание вредных серосодержащих примесей в горной породе, щебне (гравии) определяют в следующем порядке.
При наличии в горной породе, щебне (гравии) сульфатных и сульфидных соединений определяют общее содержание серы методами весового или йодометрического титрования, затем — содержание сульфатной серы и по их разности вычисляют содержание сульфидной серы. При наличии в горной породе, щебне (гравии) только сульфатных соединений общее содержание серы не определяют.
4.7.1 Определение общего содержания серы весовым методом
Весовой метод основан на разложении навески смесью азотной и соляной кислот с последующим осаждением серы в виде сульфата бария и определением массы последнего.
4.7.1.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью измерения ±0,0002 г взвешивания.
Чашки фарфоровые диаметром 15 см по ГОСТ 9147. Стаканы стеклянные вместимостью 100,200, 300, 400 мл по ГОСТ 25336.
Печь муфельная, обеспечивающая температуру нагрева 900 °С.
Тигли фарфоровые по ГОСТ 9147.
Эксикатор по ГОСТ 25336. Баня водяная.
Фильтры безвольные: «красная лента» и «синяя лента».
Кальций хлористый (хлорид кальция) по ГОСТ 450, прокаленный при температуре 700—800 °С, для заполнения эксикатора.
Кислота азотная по ГОСТ 4461.
Кислота соляная по ГОСТ 3118.
Аммиак водный по ГОСТ 3760, 10%-ный раствор.
Бария хлорид (хлорид бария) по ГОСТ 4108, 10 %-ный раствор.
Индикатор метиловый оранжевый, 0,1 %-ный раствор.
Серебро азотнокислое (нитрат серебра) по ГОСТ 1277, 1%-ный раствор.
4.7.1.2 Порядок проведения анализа
Навеску массой 0,5—2 г помещают в стеклянный стакан вместимостью 200 мл или фарфоровую чашку, смачивают несколькими каплями дистиллированной воды, добавляют 30 мл азотной кислоты, накрывают стеклом, оставляют на 10—15 мин.
После окончания реакции добавляют 10 мл соляной кислоты, перемешивают стеклянной палочкой, накрывают стеклом и ставят стакан (чашку) в водяную баню. Через 20—30 мин после прекращения выделения бурых паров оксида азота стекло снимают и выпаривают содержимое стакана или чашки досуха.
После охлаждения остаток смачивают 5—7 мл соляной кислоты и снова выпаривают досуха. Операцию повторяют 2—3 раза, доливая 50 мл горячей воды и кипятят до полного растворения солей.
Для осаждения элементов группы полуторных оксидов в раствор добавляют 2—3 капли индикатора метилового оранжевого, доливают раствор аммиака до перехода окраски индикатора из красной в желтую и появления запаха аммиака. Через 10 мин скоагулированный осадок полуторных оксидов отфильтровывают через фильтр «красная лента» в стакан вместимостью 300—400 мл. Осадок промывают теплой водой с добавлением нескольких капель раствора аммиака. К фильтрату добавляют соляную кислоту до перехода окраски раствора в розовый цвет и добавляют еще 2— 5 мл кислоты.
Фильтрат разбавляют водой до объема 200—250 мл, нагревают до кипения и вливают в один прием 10 мл горячего раствора хлорида бария, перемешивают, кипятят раствор 5—10 мин.
Раствор с выделившимся осадком ставят в теплое место на 2—3 ч, допускается оставлять раствор до следующего дня, затем осадок отфильтровывают через плотный фильтр «синяя лента» и промывают 10 раз небольшими порциями холодной воды до удаления хлорид-ионов.
Полноту удаления хлорид-ионов проверяют по реакции с нитратом серебра: несколько капель фильтрата помещают на стекло и добавляют каплю 1 %-ного раствора нитрата серебра. Отсутствие образования белого осадка свидетельствует о полноте удаления хлорид-ионов.
В фарфоровый тигель, предварительно прокаленный до постоянной массы при температуре 800—850 °С, помещают осадок с фильтром, высушивают, озоляют, избегая воспламенения фильтра, и прокаливают в открытом тигле до полного выгорания фильтра, а затем при температуре 800—850 °С в течение 30—40 мин.
После охлаждения в эксикаторе тигель с осадком взвешивают. Прокаливание повторяют до получения постоянной массы. Для определения содержания серы в использованных для анализа реактивах параллельно с анализом проводят «глухой» опыт. Количество сульфата бария, найденное «глухим» опытом, вычитают из массы сульфата бария, полученной при анализе пробы.
4.7.1.3 Обработка результатов анализа
Общее содержание серы SO3 общ , %, в пересчете на SO3 определяют по формуле
![]() , (19)
, (19)
где m — масса навески, г;
т1 — масса осадка сульфата бария, г;
т2— масса осадка сульфата бария в «глухом» опыте, г;
0,343 — коэффициент пересчета сульфата бария на SO3 .
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 5. В случае превышения анализ следует повторить до получения допустимого расхождения.
Таблица 5 В процентах
Общее содержание серы | Абсолютное допустимое расхождение | ||||||||||
До 0,5 включ. Св. 0,5 » 1,0 » » 1,0 | 0,10 0,15 0,20 |
4.7.2 Определение общего содержания серы методом йодометрического титрования
Метод основан на сжигании навески в потоке углекислого газа при температуре 1300—1350 °С, поглощении выделяющегося SO2 раствором йода и титровании раствором тиосульфата натрия избытка йода, не вошедшего в реакцию с образовавшейся сернистой кислотой.
4.7.2.1 Средства контроля и вспомогательное оборудование
Установка для определения содержания серы (рисунок 1).
Пипетки по ГОСТ 29228.
Бюретки по ГОСТ 29252.
Натрий серноватистокислый (тиосульфат натрия) 5-водный по ГОСТ 27068, 0,005 Н раствор.
Натрий углекислый (карбонат натрия) по ГОСТ 83.
Калий двухромовокислый (бихромат калия) по ГОСТ 4220, стандарт-титр.
Крахмал растворимый по ГОСТ 10163, 1 %-ный раствор.
Йод по ГОСТ 4159, 0,005 Н раствор.
Калий йодистый (йодид калия) по ГОСТ 4232.
Кислота серная по ГОСТ 4204, 0,1 Н раствор.
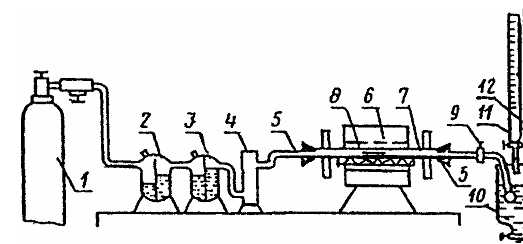
1 — баллон с углекислым газом; 2 — промывная склянка с 5 %-ным раствором сульфата меди; 3 — промывная склянка с 5 %-ным раствором перманганата калия; 4 — колонка с прокаленным хлоридом кальция; 5 — резиновые пробки; 6 — электрическая трубчатая печь с силитовыми стержнями, обеспечивающая температуру нагрева 1300 °С; 7 — фарфоровая трубка для прокаливания длиной 70—75 мм, внутренним диаметром 18—20 мм; 8 — фарфоровая лодочка № 1 (длина 70, ширина 9, высота 5—7 мм) или фарфоровая лодочка № 2 (длина 95, ширина 12, высота 10 мм) по ГОСТ 9147; 9 — кран; 10 — поглотительный сосуд; 11 — бюретка с раствором йода; 12 — бюретка с раствором тиосульфата натрия
Примечание — Все части установки соединены резиновыми трубками встык. Для предотвращения обгорания резиновых пробок внутреннюю торцевую поверхность закрывают асбестовыми прокладками.
Рисунок 1 — Схема установки для определения содержания серы
4.7.2.2 Порядок подготовки к проведению анализа
4.7.2.2.1 Приготовление 0,005 Н раствора тиосульфата натрия
Для приготовления раствора тиосульфата натрия 1,25 г Na2S2O3 ( H2O растворяют в 1 л свежепрокипяченной дистиллированной воды и прибавляют 0,1 г карбоната натрия. Раствор перемешивают и оставляют на 10—12 дн., после чего определяют его титр по 0,01 Н раствору бихромата калия, приготовленному из стандарттитра.
Для определения титра раствора тиосульфата натрия к 10 мл 0,01 Н раствора бихромата калия добавляют 50 мл 0,1 Н раствора серной кислоты, 2 г сухого йодида калия и титруют приготовленным раствором тиосульфата натрия до соломенно-желтого цвета. Затем добавляют несколько капель 1 %-ного раствора крахмала (раствор окрашивается в синий цвет) и титруют до обесцвечивания раствора.
Коэффициент поправки к титру 0,005 Н раствора тиосульфата натрия определяют по формуле
![]() , (20)
, (20)
где 10 — объем 0,005 Н раствора бихромата калия, взятого для титрования, мл;
![]() — нормальность раствора бихромата калия;
— нормальность раствора бихромата калия;
V — объем 0,005 Н раствора тиосульфата натрия, израсходованного на титрование 10 мл 0,01 Н раствора бихромата калия, мл;
![]() — нормальность раствора тиосульфата натрия.
— нормальность раствора тиосульфата натрия.
Проверку титра проводят не реже одного раза в 10 сут. Раствор тиосульфата натрия хранят в темных бутылях.
4.7.2.2.2 Приготовление 0,005 Н раствора йода
Для приготовления раствора йода 0,63 г кристаллического йода и 10 г йодида калия растворяют в 15 мл дистиллированной воды. Раствор переносят в мерную колбу вместимостью 1 л с хорошо притертой пробкой, доливают водой до метки, перемешивают и хранят в темноте.
Титр приготовленного раствора йода устанавливают по титрованному раствору тиосульфата натрия, приготовленному по 4.7.2.2.1.
10 мл 0,005 Н раствора йода титруют 0,005 Н раствором тиосульфата натрия в присутствии крахмала.
Коэффициент поправки к титру 0,005 Н раствора йода определяют по формуле
![]() , (21)
, (21)
где ![]() — объем 0,005 Н раствора тиосульфата натрия, из расходованный на титрование раствора йода, мл;
— объем 0,005 Н раствора тиосульфата натрия, из расходованный на титрование раствора йода, мл;
![]() — коэффициент поправки 0,005 Н раствора тиосульфата натрия;
— коэффициент поправки 0,005 Н раствора тиосульфата натрия;
![]() — нормальность раствора тиосульфата натрия;
— нормальность раствора тиосульфата натрия;
10 — объем раствора йода, взятого для титрования, мл;
![]() — нормальность раствора йода.
— нормальность раствора йода.
4.7.2.3 Порядок проведения анализа
Перед началом работы нагревают печь до температуры 1300 °С и проверяют герметичность установки. Для этого закрывают кран перед поглотительным сосудом и пускают углекислый газ. Прекращение прохождения пузырьков газа через промывную склянку свидетельствует о герметичности установки.
Определяют коэффициент К, устанавливающий соотношение между концентрациями раствора йода и тиосульфата натрия. Через установку пропускают углекислый газ в течение 3—5 мин, наполняют поглотительный сосуд на 2/3 водой. Из бюретки наливают 10 мл титрованного раствора йода, добавляют 5 мл 1 %-ного раствора крахмала и титруют раствором тиосульфата натрия до обесцвечивания раствора. Коэффициент соотношения концентраций растворов йода и тиосульфата натрия К равен среднему значению из трех определений. Коэффициент соотношения концентраций К в лабораторных условиях определяют ежедневно перед испытаниями.
Навеску массой 0,1—1,0 г помещают в предварительно прокаленную лодочку. В поглотительный сосуд заливают 250—300 мл дистиллированной воды, добавляют отмеренный бюреткой объем раствора йода и 5 мл раствора крахмала и перемешивают потоком углекислого газа.
Лодочку с навеской с помощью крючка из жаростойкой проволоки помещают в разогретую трубку (со стороны подачи углекислого газа). Закрывают трубку пробкой и подают углекислый газ (скорость подачи 90—100 пузырьков в 1 мин).
Навеску прокаливают 10—15 мин, следя за тем, чтобы раствор в поглотительном сосуде сохранял синюю окраску. Затем раствор в поглотительном сосуде фильтруют раствором тиосульфата натрия до обесцвечивания раствора. После окончания титрования извлекают лодочку из печи, стараясь не загрязнить стенки фарфоровой трубки остатками навески.
В поглотительный сосуд, промытый водой, наливают новую порцию воды, раствора йода и крахмала.
4.7.2.4 Обработка результатов анализа
Массовую долю общего содержания серы SO3 общ , %, в пересчете на SO3 определяют по формуле
![]() , (22)
, (22)
где V — объем раствора йода, взятого для титрования, мл;
К — коэффициент соотношения концентраций раствора йода и тиосульфата натрия;
V1 — объем раствора тиосульфата натрия, израсходованного на титрование избытка йода, не вступившего в реакцию, мл;
т — масса навески, г;
![]() / SO3 — титр 0,005 Н раствора йода по SO3, определяется по формуле
/ SO3 — титр 0,005 Н раствора йода по SO3, определяется по формуле
![]() / SO3 =
/ SO3 =![]() 0,0002, (23)
0,0002, (23)
где ![]() — коэффициент поправки к титру 0,005 Н раствора йода, рассчитанный по формуле 21;
— коэффициент поправки к титру 0,005 Н раствора йода, рассчитанный по формуле 21;
0,0002 — количество SO3 соответствующее 1 мл 0,005 Н раствора йода, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 6. В случае превышения расхождения опыт следует повторить до получения допустимого расхождения.
Таблица 6 В процентах
Массовая доля SO3 | Абсолютное допустимое расхождение | ||||||||||
До 0,5 включ. Св. 0,5 » 1,0 » » 1,0 | 0,05 0,15 0,20 |
4.7.3 Определение сульфатной серы
Метод основан на разложении навески соляной кислотой с последующим осаждением серы в виде сульфата бария и определении массы последнего.
4.7.3.1 Средства контроля и вспомогательное оборудование
Для проведения анализа применяют аппаратуру, реактивы и растворы, указанные в 4.7.2.1, а также соляную кислоту по ГОСТ 3118 (раствор 1:3).
4.7.3.2 Порядок проведения анализа
Навеску массой 1 г помещают в стакан вместимостью 100— 150 мл, прикрывают стеклом и добавляют 40—50 мл соляной кислоты. После прекращения выделения пузырьков газа ставят стакан на плитку и выдерживают при слабом кипении 10—15 мин. Осаждают полуторные оксиды, добавляя 2—3 капли индикатора метилового оранжевого и доливая раствор аммиака до перехода окраски индикатора из красной в желтую и появления запаха аммиака. Через 10 мин осадок отфильтровывают. Осадок промывают теплой водой с добавлением нескольких капель раствора аммиака.
Фильтрат нейтрализуют соляной кислотой до перехода окраски раствора в розовую и доливают еще 2,5 мл кислоты. Раствор нагревают до кипения и добавляют в один прием 10 мл горячего раствора хлорида бария, перемешивают, кипятят раствор 5—10 мин и оставляют раствор с выделившимся осадком на 2—3 ч (допускается до следующего дня). Осадок отфильтровывают через плотный фильтр «синяя лента» и промывают 10 раз небольшими порциями холодной воды до удаления хлорид-ионов.
Полноту удаления хлорид-ионов проверяют по реакции с нитратом серебра: несколько капель фильтрата помещают на стекло и добавляют каплю 1 %-ного раствора нитрата серебра. Отсутствие образования белого осадка свидетельствует о полноте удаления хлорид-ионов.
В фарфоровый тигель, предварительно прокаленный до постоянной массы при температуре 800—850 °С, помещают осадок с фильтром, высушивают, озоляют, избегая воспламенения фильтра, и прокаливают в открытом тигле до полного выгорания фильтра, а затем при температуре 800—850 °С в течение 30—40 мин.
После охлаждения в эксикаторе тигель с осадком взвешивают. Прокаливание повторяют до получения постоянной массы.
Параллельно с анализом проводят «глухой» опыт. Количество сульфата бария, найденное «глухим» опытом, вычитают из массы сульфата бария, полученного при анализе пробы.
4.7.3.3 Обработка результатов анализа
Содержание сульфатной серы SO3 cульфат , %, в пересчете на SО3, определяют по формуле
![]() , (24)
, (24)
где т1 — масса осадка сульфата бария, г;
т2 — масса осадка сульфата бария в «глухом» опыте, г;
0,343 — коэффициент пересчета сульфата бария на SO3
т — масса навески, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 7. В случае превышения расхождения опыт следует повторить до получения допустимого расхождения.
Таблица 7 В процентах
Массовая доля SO3 сульфат | Абсолютное допустимое расхождение | ||||||||||
До 0,5 включ. Св. 0,5 » 1,0 » » 1,0 | 0,10 0,15 0,20 |
4.7.4 Определение сульфидной серы
Содержание сульфидной серы SO3 сульфид определяют по разности между общим содержанием серы (4.7.1) и содержанием сульфатной серы (4.7.3). .
4.7.4.1 Обработка результатов анализа
Содержание сульфидной серы SO3 сульфид, %, в пересчете на SО3 определяют по формуле
SO3 сульфид = SO3 общ — SO3 сульфат, (25)
где SO3 общ — общее содержание серы в пересчете на SO3 ,%;
SO3сульфат— содержание сульфатной серы в пересчете на SO3, %.
Содержание сульфидной серы S судьфид, %, в пересчете на S определяют по формуле
S судьфид = 0,4 ( SO3судьфид, (26)
где 0,4 - коэффициент пересчета SO3судьфид на S судьфид ;
SO3судьфид — содержание сульфидной серы, определенное по формуле 25.
4.8 Определение оксидов калия и натрия
Метод основан на измерении интенсивности излучения линий элементов, образующихся в пламени горящих газов (пропан-бутановой смеси) и воздуха при введении в него анализируемого раствора и растворов сравнения. Интенсивность излучения линии натрия измеряют при длине волны 590 нм, калия — при 770 нм. Для расчета содержания натрия и калия пользуются градуировочными графиками. Присутствие в растворе алюминия, железа, магния не влияет на определение содержания щелочных оксидов. Влияние кальция на определение натрия устраняют введением в эталонные растворы хлорида кальция.
4.8.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104.
Фотометр пламенный, работающий на пропан-бутановой смеси.
Печь муфельная, обеспечивающая температуру (1000±50) °С.
Баня водяная или песчаная.
Колбы мерные по ГОСТ 1770 вместимостью 100 мл и 1 л.
Чашки платиновые по ГОСТ 6563.
Стаканы вместимостью 50 мл по ГОСТ 19908.
Кислота серная по ГОСТ 4204 плотностью 1,84.
Кислота фтористоводородная (плавиковая) по ГОСТ 10484,
40 %-ный раствор.
Натрий сернокислый (сульфат натрия) по ГОСТ 4166.
Калий сернокислый (сульфат калия) по ГОСТ 4145.
Кислота соляная по ГОСТ 3118, раствор 1:5.
Кальций углекислый (карбонат кальция) по ГОСТ 4530.
Типовой раствор натрия и калия (раствор А), содержащий 2,0 г Na2O и 2,0 г К2О в 1 л, 4,583 г Na2SO4 и 3,7 г K2SO4 растворяют в мерной колбе вместимостью 1 л.
Типовой раствор кальция (раствор Б), содержащий 0,25 г СаО в 1 л, 0,4555 г высушенного углекислого кальция (карбоната кальция) помещают в мерную колбу вместимостью 1 л, добавляют 200 мл воды и осторожно небольшими дозами прибавляют 45—50 мл соляной кислоты (1:5). Затем содержимое колбы доводят водой до метки.
Приготовление эталонных растворов и построение градуировочных графиков
При определении натрия и калия на пламенном фотометре фотометрируют серию эталонных растворов с известным содержанием натрия и калия, в которых количество кальция почти такое же, как в анализируемом материале. Если оксида кальция в пробе не более 1 %, для приготовления эталонных растворов используют типовой раствор, не содержащий кальций (раствор А).
В пять колб вместимостью 1 л наливают раствор А в количестве, указанном в таблице 8, и добавляют дистиллированной воды до метки.
Эти растворы фотометрируют и на основании полученных данных строят градуировочный график № 1, откладывая по оси абсцисс содержание Na2O (соответственно К2О), по оси ординат — показание стрелки гальванометра.
Состав эталонных растворов, применяемых для построения градуировочного графика для проб, не содержащих СаО, указан в таблице 8.
Таблица 8
Номер эталонного раствора | Содержание, мг/л | Количество раствора А, мл | |
Na2O | К2О | ||
1 | 200 | 200 | 100 |
2 | 150 | 150 | 75 |
3 | 100 | 100 | 50 |
4 | 50 | 50 | 25 |
5 | 20 | 20 | 10 |
При анализе материалов, содержащих СаО более 1 %, пользуются градуировочными графиками № 2—5, построенными на основании результатов фотометрирования эталонных растворов, в состав которых входит кальций. Эталонные растворы в этом случае готовят следующим образом. В мерные колбы вместимостью 1 л вводят растворы А и Б в количестве от 5,0 до 60,0 мл. Затем к содержимому колбы добавляют дистиллированной воды до метки.
Таким способом приготавливают четыре серии эталонных растворов. Каждая серия состоит из пяти эталонных растворов с различным содержанием Na2O и К2О, но с одинаковым содержанием СаО.
Содержание СаО в эталонных растворах в зависимости от содержания СаО в пробе должно соответствовать значениям, указанным в таблице 9.
Таблица 9
x | Содержание СаО в эталонных растворах, мг/л | Количество раствора Б, мл | Содержание СаО в анализируемой пробе, % | Масса навески, г |
1 | 0 | 0 | (1 | Не нормируется |
2 | 50 | 5 | Св. 1 до 3 » 3 » 5 | 0,2 0,1 |
3 | 100 | 10 | » 5 » 7 » 7 » 10 | 0,2 0,1 |
4 | 300 | 30 | » 10 » 20 » 20 » 30 | 0,2 0,1 |
5 | 600 | 60 | » 30 » 40 » 40 » 50 50 и более | 0,2 0,15 0,1 |
4.8.2 Порядок проведения анализа
Навеску массой 0,1—0,2 г разлагают в платиновой чашке смесью 5 мл серной и 10—20 мл фтористоводородной кислоты сначала при медленном, затем при сильном нагревании. После этого содержимое чашки выпаривают досуха, пока не удалятся фтористоводородная и серная кислоты. После этого чашку ставят в муфельную печь, нагретую до 600 °С.
Через 10—15 мин чашку вынимают из муфеля, остаток в чашке растворяют в небольшом количестве дистиллированной воды и фильтруют в мерную колбу вместимостью 100 мл.
Осадок, образовавшийся на фильтре, промывают теплой водой, объем содержимого колбы доводят водой до метки, затем наливают в стаканчик вместимостью 50 мл и определяют на пламенном фотометре натрий и калий в соответствии с инструкцией к прибору.
4.8.3 Обработка результатов анализа
Содержание оксида калия находят по градуировочному графику № 1. При определении оксида натрия пользуются графиком, который построен по результатам фотометрирования растворов, содержащих кальция столько же, сколько и анализируемый раствор, при этом учитывают массу взятой навески.
Массовую долю оксидов натрия Na2O и калия К2О, %, определяют по формуле
![]() , (27)
, (27)
где С — количество Na2O или К2О, определенное по градуировочному графику, мг/л;
V — общий объем анализируемого раствора, мл;
m — масса навески, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 10.
Таблица 10 В процентах
Массовая доля оксида натрия (калия) | Абсолютное допустимое расхождение |
До 1,0 включ. Св. 1,0 » 5,0 » » 5,0 | 0,05 0,10 0,25 |
4.9 Определение оксида железа двухвалентного
Метод основан на кислотном разложении анализируемой пробы в потоке углекислого газа с последующим титрованием двухвалентного железа перманганатом калия.
4.9.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью измерения не более 0,0002 г.
Электроплитка с закрытой спиралью.
Баня песчаная.
Аппарат Киппа, в котором получают углекислый газ действием раствора соляной кислоты на мраморную крошку или известняк.
Колбы конические по ГОСТ 25336 вместимостью 250 мл.
Бюретки по ГОСТ 29251 и ГОСТ 29252.
Тигель платиновый по ГОСТ 6563.
Кислота серная по ГОСТ 4204, раствор 1:4.
Калий марганцовокислый (перманганат калия) по ГОСТ 20490 0,1 Н титрованный раствор (приготавливают из стандарт-титра).
Кислота соляная по ГОСТ 3118, раствор 1:3.
Мраморная крошка или известняк.
Кислота фтористоводородная (плавиковая) по ГОСТ 10484.
4.9.2 Порядок проведения анализа
4.9.2.1 В коническую колбу вместимостью 250 мл наливают 100 мл серной кислоты. Колбу закрывают пробкой с двумя отверстиями, в которые вставлены стеклянные трубки, согнутые под прямым углом. Одна из трубок (по ходу газа) доходит до дна колбы, вторая кончается под пробкой. Длинную трубку присоединяют к аппарату Киппа с углекислым газом. Открыв кран у аппарата, пропускают поток углекислого газа через колбу в течение 3 мин. В это время отвешивают на сухом часовом стекле 1—1,5 г навески материала, находящегося в воздушно-сухом состоянии. Приоткрыв пробку, быстро всыпают навеску в колбу. Не прекращая тока газа, взвешивают стекло и по разности определяют массу навески. Содержимое колбы кипятят 30 мин, пропуская при этом ток углекислого газа. Затем снимают колбу с плитки и, не прекращая тока газа, охлаждают содержимое колбы. После чего отсоединяют колбу от прибора Киппа, прибавляют 150 мл свежепрокипяченной холодной воды и титруют 0,1 Н раствором марганцовокислого калия (перманганатом калия) до розового цвета, не исчезающего в течение 20—30 с.
4.9.2.2 Материалы, не растворяющиеся в серной кислоте без остатка, разлагают в смеси фтористоводородной (плавиковой) и серной кислот.
Навеску анализируемого материала в воздушно-сухом состоянии массой 0,5—1 г помещают в платиновый тигель, смачивают водой, прибавляют 10 мл раствора серной кислоты, доливают до половины тигля горячую свежепрокипяченную дистиллированную воду, закрывают тигель крышкой с отверстием, вставляют в него стеклянную трубку от аппарата Киппа и пропускают углекислый газ. Тигель нагревают на песчаной бане, пропуская углекислый газ, до начала кипения жидкости. Затем прекращают подачу углекислого газа (отсоединяют от прибора), приоткрывают крышку и быстро прибавляют 7 мл фтористоводородной кислоты.
Тигель плотно закрывают крышкой (без отверстия) и осторожно нагревают до появления белых паров. После чего содержимое тигля кипятят 10 мин.
Содержимое тигля переносят в стакан вместимостью 400— 500 мл, прибавляют 150 мл свежепрокипяченной и охлажденной до комнатной температуры воды, 5 мл серной кислоты и быстро титруют 0,1 Н раствором марганцовокислого калия до розового цвета, не исчезающего в течение 20—30 с.
4.9.3 Обработка результатов анализа
Массовую долю оксида железа (II) FeO, %, определяют по формуле
![]() , (28)
, (28)
где V — объем 0,1 Н раствора марганцовокислого калия (перманганата калия), пошедшего на титрование, мл;
0,007184 — количество оксида железа, соответствующее 1 мл точно 0,1 Н раствора марганцовокислого калия (перманганата калия), г;
т — масса исходной навески, пересчитанная на высушенную при 105 °С, г.
Пересчет массы навески на высушенную производят по формуле (1). Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 11.
Таблица 11 В процентах
Массовая доля оксида железа (II) | Абсолютное допустимое расхождение |
До 1,0 включ. От 1,0 » 5,0 » » 5,0 » 10,0 | 0,08 0,25 0,50 |
4.10 Определение общего содержания хлоридов и легкорастворимых хлоридов
Общее содержание хлоридов в щебне (гравии) определяют осаждением С1( в азотнокислой среде избытком нитрата серебра. Избыток нитрата серебра оттитровывают роданидом аммония или калия в присутствии индикатора — железоаммонийных квасцов. В момент окончания осаждения хлорида серебра (достижения эквивалентной точки) роданид аммония образует роданид железа, окрашивающий раствор в красный цвет.
Легкорастворимыми соединениями хлора условно считают хлориды, переходящие в водную вытяжку из пробы щебня (гравия) при обработке ее водой.
4.10.1 Определение общего содержания хлоридов
4.10.1.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104 с погрешностью взвешивания 0,0002 г.
Шкаф сушильный.
Электроплитка с закрытой спиралью.
Кислота азотная по ГОСТ 4461, раствор 1:40 и 6 М.
Квасцы железоаммонийные, 40 %-ный насыщенный раствор.
Аммоний роданистый (тиоцианат аммония) по ГОСТ 27067 или калий роданистый (тиоцианат калил) по ГОСТ 4139, 0,1 М титрованный раствор.
Натрий хлористый (хлорид натрия) по ГОСТ 4233, 0,1 М раствор.
Калий хромовокислый (хромат калия) по ГОСТ 4459, 10 %-ный раствор.
Серебро азотнокислое (нитрат серебра) по ГОСТ 1277 — 0,1 М титрованный раствор.
Нитробензол.
4.10.1.2 Порядок подготовки к проведению анализа
Титр раствора нитрата серебра 0,1 М устанавливают по хлориду натрия. Для этого отбирают 10 мл точно 0,1 М раствора хлорида натрия и титруют AgNO3 в присутствии 1 мл 10 %-ного раствора K2CrO4. Титр раствора нитрата серебра, выраженный в г/мл Сl(, определяют по формуле
![]() , (29)
, (29)
где V — количество точно 0,1 М раствора NaCl, израсходованное на титрование, мл;
V1— количество раствора AgNO3, израсходованное на титрование, мл;
0,00355 — количество Cl-, соответствующее 1 мл точно 0,1 М раствора NaCl, г.
Перед титрованием определяют коэффициент К, выражающий соотношение между концентрациями растворов AgNO3 и NH4CNS.
Для этого берут 10 мл раствора AgNO3, прибавляют 5 мл 6 М HNO3 и 1 мл раствора железоаммонийных квасцов и титруют 0,1 М раствором роданида аммония. До эквивалентной точки цвет становится красновато-коричневым. Титрование продолжают до сохраняющейся после сильного встряхивания окраски.
Коэффициент К определяют как средний результат пяти титровании по формуле
![]() , (30)
, (30)
где V — количество раствора NH4CNS, израсходованное на титрование 10 мл раствора AgNO3.
4.10.1.3 Порядок проведения анализа
Навеску массой 0,7—1,5 г помещают в стакан вместимостью 100— 150 мл и при медленном нагревании обрабатывают 30 мл HNO3 (раствор 1:40). После прекращения выделения пузырьков газа раствор нагревают 5—10 мин, затем фильтруют через плотный фильтр «синяя лента», промывают 5—6 раз горячей водой. Фильтрат собирают в колбу, в которой титруют хлориды.
Титрование производят следующим образом. К фильтрату добавляют 5 мл 6 М НNО3 и 1 мл нитробензола и постепенно, энергично помешивая, приливают из бюретки избыточное количество AgNO3.
Содержание колбы взбалтывают до тех пор, пока осадок не соберется в хлопья. Затем прибавляют 1 мл (15—20 капель) раствора железоаммонийных квасцов и титруют раствором NH4CNS, энергично помешивая после каждой капли, если при осторожном помешивании цвет исчезает. Раствор NH4CNS прибавляют до слабой окраски (красновато-коричневой), не исчезающей при слабом помешивании. Полностью окрашенный раствор взбалтывают осторожно, так как после точки эквивалентности при энергичном взбалтывании возможно обесцвечивание раствора за счет частичного растворения осадка хлорида серебра.
4.10.1.4 Обработка результатов анализа Массовую долю хлоридов, %, определяют по формуле
![]() , (31)
, (31)
где V1 — количество раствора нитрата серебра, добавленное до титрования, мл;
V2 — количество раствора роданида аммония, израсходованное на обратное титрование, мл;
К — коэффициент, выражающий соотношение между концентрациями раствора нитрата серебра и роданида аммония;
TCl— титр раствора нитрата серебра по Сl(, г/мл;
т — масса сухой навески, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 12.
Таблица 12 В процентах
Массовая доля С1( | Абсолютное допустимое расхождение |
До 0,1 включ. | 0,02 |
Св. 0,1 » 0,5 » | 0,03 |
» 0,5 » 1,0 » | 0,05 |
» 1,0 » 3,0 » | 0,07 |
» 3,0 » 10,0 » | 0,15 |
» 10,0 » 25,0 » | 0,4 |
Более 25,0 | 0,6 |
4.10.2 Определение легкорастворимых хлоридов
Для проведения анализа приготавливают водную вытяжку состава 1:10. Для этого точную навеску массой 5 г помещают в коническую колбу и приливают в нее пипеткой 50 мл свежепрокипяченной дистиллированной воды, не содержащей углекислоты. Содержимое колбы интенсивно перемешивают и через 10 мин фильтруют через неплотный фильтр «белая лента». Остаток в колбе промывают три раза небольшими порциями воды.
Фильтрат и промывные воды собирают в коническую колбу, добавляют в нее 5 мл 6 М HNO3, перемешивают, приливают из бюретки избыточное количество AgNO3 и далее проводят титрование как указано в 4.10.1.3. Массовую долю легкорастворимых хлоридов рассчитывают по формуле (31).
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 12.
4.11 Определение оксида марганца
Метод основан на окислении ионов двухвалентного марганца в азотнокислой среде персульфатом аммония до семивалентных перманганат-ионов, окрашенных в фиолетовый цвет в присутствии ионов серебра.
Оптическую плотность раствора перманганата измеряют на фотоэлектроколориметре с зеленым светофильтром или на спектрофотометре при длине волны 530 нм.
4.11.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104.
Фотоэлектроколориметр или спектрофотометр.
Печь муфельная.
Баня водяная.
Тигель платиновый по ГОСТ 6563.
Колбы мерные вместимостью 1 л, 200 мл, 100 мл по ГОСТ 1770. Пипетки вместимостью 10, 25 и 50 мл по ГОСТ 29227 и ГОСТ 29228.
Стаканы вместимостью 100—150 мл по ГОСТ 25336. Натрий углекислый (карбонат натрия) по ГОСТ 83.
Калий углекислый (карбонат калия) по ГОСТ 4221.
Натрий тетраборнокислый (тетраборат натрия) по ГОСТ 4199 (безводный).
Калий азотнокислый (нитрат калия) по ГОСТ 4217 или аммоний азотнокислый (нитрат аммония) по ГОСТ 22867.
Смесь для сплавления: натрий углекислый (карбонат натрия), натрий тетраборнокислый (тетраборат натрия) смешивают в соотношении 2:1 или натрий углекислый (карбонат натрия), калий углекислый (карбонат калия) и натрий тетраборнокислый (тетраборат натрия) смешивают в соотношении 1:1:1. Для полноты окисления низковалентных форм железа, серы, марганца и т.п. в смесь для сплавления можно добавить 0,5 % по массе азотнокислого калия или 1 % по массе азотнокислого аммония, обеспечивая во избежание порчи платиновых тиглей равномерное распределение соли по всей массе плавня.
Водорода пероксид по ГОСТ 10929 — раствор 1:10 или кислота аскорбиновая пищевая.
Кислота азотная по ГОСТ 4461, растворы 1:4 и 1 М.
Кислота ортофосфорная по ГОСТ 6552.
Серебро азотнокислое (нитрат серебра) по ГОСТ 1277, раствор, содержащий 2 г/л.
Калий марганцовокислый (перманганат калия) по ГОСТ 20490 или стандарт-титр — раствор 0,1 Н.
Аммоний надсернокислый (персульфат аммония) по ГОСТ 20478.
4.11.2 Порядок подготовки к проведению анализа
Готовят стандартный раствор А, растворяя 0,1 Н стандарт-титр марганцовокислого калия (перманганата калия) в 1 л воды. 1 мл раствора А содержит 1,419 мг МnО.
Готовят градуировочный раствор Б: 14,1 мл раствора А разбавляют в мерной колбе вместимостью 200 мл до метки водой и хорошо перемешивают. Полученный градуировочный раствор содержит 0,1 г/мл МnО.
Затем строят градуировочный график.
В пять мерных колб вместимостью 100 мл приливают соответственно 2,5; 5,0; 7,5; 10,0; 15,0 мл раствора Б, что соответствует 0,25; 0,50; 0,75; 1,00 и 1,5 мг МnО, разбавляют до метки 1 М раствором азотной кислоты, затем перемешивают и фотометрируют на фотоколориметре с зеленым светофильтром или на спектрофотометре при длине волны 530 нм, используя кювету с толщиной поглощающего свет слоя 10 мм.
По полученным результатам определений оптической плотности и известной концентрации оксида марганца (II) в фотометрируемых растворах строят градуировочный график.
4.11.3 Порядок проведения анализа
Навеску массой 0,1—0,5 г сплавляют с 2 г смеси для сплавления. Плав растворяют в 50 мл теплого раствора азотной кислоты 1:4 в стакане вместимостью 150 мл. Тигель обмывают минимальным количеством воды и переносят раствор в мерную колбу вместимостью 100—200 мл. Если выделяются бурые хлопья диоксида марганца, к анализируемому раствору добавляют 1—2 капли пероксида водорода или кристаллик аскорбиновой кислоты. Затем прозрачный раствор доводят до метки водой и перемешивают.
В мерную колбу вместимостью 100 мл отбирают аликвотную часть в количестве 10—50 мл анализируемого раствора, прибавляют 5 мл ортофосфорной кислоты, 5 мл азотнокислого серебра (нитрата серебра), 2—3 г надсернокислого аммония (персульфата аммония) и нагревают колбу несколько минут на водяной бане до установления интенсивной окраски, затем охлаждают, доводят водой до метки и фотометрируют.
4.11.4 Обработка результатов анализа
Массовую долю оксида марганца МnО в исследуемом материале, %, определяют по формуле
![]() , (32)
, (32)
где V — объем окрашенного исследуемого раствора, мл;
Vo — общий объем исследуемого раствора, мл;
т — масса навески, г;
Va — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл;
Сx — концентрация оксида марганца в окрашенном исследуемом растворе, мг/мл, найденная по градуировочному графику или методом сравнения по формуле
![]() , (33)
, (33)
где Сc — концентрация раствора сравнения, г/мл;
Дх — оптическая плотность окрашенного исследуемого раствора;
Дc — оптическая плотность раствора сравнения.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 13.
Таблица 13 В процентах
Массовая доля оксида марганца(II) | Абсолютное допустимое расхождение |
До 0,5 включ. | 0,04 |
От 0,5 » 1,0 » | 0,05 |
» 1,0 » 2,0 » | 0,10 |
» 2,0 » 5,0 » | 0,20 |
» 5,0 » 20,0 » | 0,60 |
4.12 Определение диоксида титана
Метод основан на образовании в сернокислой среде окрашенного в желтый цвет комплексного соединения титана с пероксидом водорода.
Оптическую плотность раствора определяют на фотоэлектроко лориметре с синим светофильтром или на спектрофотометре при длине волны 410 нм.
4.12.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104.
Печь муфельная.
Тигель платиновый по ГОСТ 6563.
Колбы мерные вместимостью на 100,250 и 500 мл по ГОСТ 1770.
Пипетки на 5, 10, 15, 20 и 25 мл по ГОСТ 29227 и ГОСТ 29228.
Шкаф сушильный.
Фотоэлектроколориметр или спектрофотометр.
Смесь для сплавления по 4.11.1.
Натрий углекислый (карбонат натрия) по ГОСТ 83.
Калий углекислый (карбонат калия) по ГОСТ 4221.
Аммоний азотнокислый (нитрат аммония) по ГОСТ 22867.
Натрий тетраборнокислый (тетраборат натрия) по ГОСТ 4199, обезвоженный при температуре (400±20) °С.
Кислота соляная по ГОСТ 3118, раствор 1:3.
Кислота серная по ГОСТ 4204, раствор 1:20.
Кислота ортофосфорная по ГОСТ 6552.
Водорода пероксид по ГОСТ 10929, раствор 1:10.
Титана (IV) диоксид по действующей технической документации.
4.12.2 Порядок подготовки к проведению анализа
Приготовление стандартного раствора
Навеску диоксида титана (IV) массой 0,05 г сплавляют в платиновом тигле с 1 г смеси для сплавления по 4.11.1 при температуре 900—950 °С в течение 10 мин. Плав растворяют в 100 мл раствора соляной кислоты, количественно переносят в мерную колбу вместимостью 500 мл, доливают до метки водой и перемешивают. Массовая концентрация стандартного раствора диоксида титана — 0,1 мг/мл.
Построение градуировочного графика.
В пять мерных колб вместимостью по 100 мл приливают 2,5; 5; 10; 15; 20 мл стандартного раствора, что соответствует 0,25; 0,50; 1,00; 1,50; 2,00 мг диоксида титана.
В каждую колбу добавляют по 10 мл раствора соляной кислоты, 3— 5 капель ортофосфорной кислоты, доводят объем примерно до 50 мл водой и добавляют по 3 мл раствора пероксида водорода, доливают до метки раствором серной кислоты, тщательно перемешивают и полученные градуировочные растворы фотометрируют относительно дистиллированной воды на фотоэлектроколориметре с синим светофильтром или на спектрофотометре при длине волны 410 нм, используя кювету с толщиной слоя, поглощающего свет, 50 мм.
По полученным результатам определений оптической плотности и известной концентрации диоксида титана в фотометрируемых растворах строят градуировочный график.
4.12.3 Порядок проведения анализа
Навеску массой 0,5 г помещают в платиновый тигель, смешивают с 2 г смеси для сплавления и сплавляют при температуре 900— 950 °С в течение 10—20 мин.
Плав обрабатывают 50 мл раствора соляной кислоты. Полученный раствор переводят в мерную колбу вместимостью 250 мл, доливают до метки раствором серной кислоты и перемешивают.
Из полученного раствора отбирают аликвотную часть объемом 25— 50 мл в зависимости от предполагаемой массовой доли диоксида титана в анализируемой пробе и переносят в мерную колбу вместимостью 100 мл, добавляют 3 мл пероксида водорода, затем доливают до метки раствором серной кислоты, перемешивают и фотометрируют полученный раствор на фотоэлектроколориметре с синим светофильтром или на спектрофотометре при длине волны 410 нм, используя кювету с толщиной слоя, поглощающего свет, 50 мм.
4.12.4 Обработка результатов анализа
Массовую долю диоксида титана в исследуемом материале, %, определяют по формуле
![]() , (34)
, (34)
где V — объем окрашенного исследуемого раствора, мл;
vo— общий объем исследуемого раствора, мл;
т — масса навески, г;
Va — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл;
Сx— концентрация диоксида титана в окрашенном исследуемом растворе, мг/мл, найденная по градуировочному графику или методом сравнения по формуле
![]() , (35)
, (35)
где Сc — концентрация раствора сравнения, мг/мл;
Дx — оптическая плотность окрашенного исследуемого раствора;
Дc — оптическая плотность раствора сравнения.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 14.
Таблица 14 В процентах
Массовая доля диоксида титана | Абсолютное допустимое расхождение |
До 0,5 включ. Св. 0,5 » 1,5 » » 1,5 » 5,0 » | 0,06 0,10 0,30 |
4.13 Определение оксида хрома
Метод основан на взаимодействии дифенилкарбазида с ионами шестивалентного хрома с образованием в кислой среде соединения, окрашенного в красно-фиолетовый цвет.
Оптическую плотность раствора определяют на фотоэлектроколориметре с зеленым светофильтром или на спектрофотометре при длине волны 540 нм.
4.13.1 Средства контроля и вспомогательное оборудование
Весы лабораторные по ГОСТ 24104.
Фотоэлектроколориметр или спектрофотометр.
Печь муфельная.
Тигель платиновый по ГОСТ 6563.
Колбы мерные по ГОСТ 1770 вместимостью 100 и 250 мл.
Пипетки по ГОСТ 29227 и по ГОСТ 29228 на 2,5, 10, 20, 25 и 50 мл.
Стаканы по ГОСТ 25336 на 100—200 мл.
Кислота соляная по ГОСТ 3118, раствор 1:5.
Кислота ортофосфорная по ГОСТ 6552, раствор 1:2.
Спирт этиловый ректификованный технический по ГОСТ 18300.
Дифенилкарбазид: 0,25 г дифенилкарбазида растворяют при слабом нагревании в 100 мл раствора этилового спирта 1:1. Дифенилкарбазид нестоек и его раствор готовят непосредственно перед определением.
Калий двухромовокислый (бихромат калия) по ГОСТ 4220, перекристаллизованный и высушенный до постоянной массы при температуре 130 °С.
Натрий углекислый (карбонат натрия) по ГОСТ 83.
Калий углекислый (карбонат калия) по ГОСТ 4221.
Аммоний азотнокислый (нитрат аммония) по ГОСТ 22867.
Натрий тетраборнокислый (тетраборат натрия) 10-водный по ГОСТ 4199, обезвоженный при температуре (400 ± 20) °С.
Смесь для сплавления по 4.11.1.
4.13.2 Порядок подготовки к анализу
Приготовление стандартного раствора
Навеску двухромовокислого калия (бихромата калия) массой 0,0484 г растворяют в 100 мл воды. Раствор переносят в мерную колбу вместимостью 250 мл, разбавляют до метки водой и тщательно перемешивают.
Аликвотную часть этого раствора объемом 25—50 мл разбавляют водой до метки в мерной колбе вместимостью 250 мл. Массовая концентрация оксида хрома в стандартном растворе — 0,02 мг/мл.
Построение градуировочного графика
В пять мерных колб вместимостью по 100 мл наливают соответственно 0,4; 0,8; 1,2; 1,6; 2,0 мл стандартного раствора, что соответствует 0,008; 0,016; 0,024; 0,032; 0,040 г оксида хрома, разбавляют примерно до 70 мл водой, добавляют в каждую колбу по 1,5 мл соляной кислоты, 4 мл ортофосфорной кислоты, тщательно перемешивают, затем прибавляют по 1,5 мл дифенилкарбазида, разбавляют до метки водой, вновь перемешивают и через 5—7 мин фотометрируют полученные градуировочные растворы относительно дистиллированной воды на фотоэлектроколориметре с зеленым светофильтром или спектрофотометре при длине волны 540 нм, используя кювету с толщиной слоя, поглощающего свет, 30 мм. По полученным результатам определений оптической плотности и известной концентрации оксида хрома в фотометрируемых растворах строят градуировочный график.
4.13.3 Порядок проведения анализа
Навеску массой 0,2—0,5 г (в зависимости от предполагаемой массовой доли оксида хрома) сплавляют с 2 г смеси для сплавления. После охлаждения плав растворяют в 40—50 мл раствора соляной кислоты, переводят раствор в мерную колбу вместимостью 100 мл, разбавляют до метки водой и тщательно перемешивают.
Для определения массовой доли оксида хрома отбирают аликвотную часть полученного раствора объемом 10—25 мл (в зависимости от предполагаемой массовой доли элемента) в мерную колбу вместимостью 100 мл, добавляют 50—60 мл воды, 4 мл раствора ортофосфорной кислоты и перемешивают. Далее прибавляют 1,5 мл дифенилкарбазида, разбавляют до метки водой, перемешивают и через 5—7 мин фотометрируют полученный раствор на фотоэлектроколориметре с зеленым светофильтром или на спектрофотометре при длине волны 540 нм, используя кювету с толщиной слоя, поглощающего свет, 30 мм.
4.13.4 Обработка результатов анализа
Массовую долю оксида хрома в исследуемом материале, %, определяют по формуле
![]() , (36)
, (36)
где V — объем окрашенного исследуемого раствора, мл;
Vo — общий объем исследуемого раствора, мл;
т — масса навески, г;
Va — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл;
Сx — концентрация оксида хрома в окрашенном исследуемом растворе, мг/мл, найденная по градуировочному графику или методом сравнения по формуле
![]() , (37)
, (37)
где Сc — концентрация раствора сравнения, мг/мл;
Дx — оптическая плотность окрашенного исследуемого раствора;
Дc — оптическая плотность раствора сравнения.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 15.
Таблица 15 В процентах
Массовая доля оксида хрома | Абсолютное допустимое расхождение |
До 0,3 включ. Св. 0,3 » 1,0 » » 1,0 » 3,0 » » 3,0 » 8,0 » | 0,03 0,10 0,25 0,50 |
4.14 Определение оксида фосфора
Метод основан на получении фосфорномолибденовой гетерополикислоты, которая в кислой среде при восстановлении сернокислым гидразином окрашивает раствор в синий цвет.
Оптическую плотность окрашенного в синий цвет раствора определяют на фотоэлектроколориметре с красным светофильтром с максимальным пропусканием при длине волны 600—750 нм или на спектрофотометре при длине волны 830 нм.
4.14.1 Средства контроля и вспомогательное оборудование
Весы лабораторные общего назначения по ГОСТ 24104.
Фотоэлектроколориметр или спектрофотометр.
Печь муфельная.
Колбы мерные по ГОСТ 1770 вместимостью 50, 100, 200—250, 500мл.
Пипетки вместимостью 1,5; 5; 10; 20; 25 мл по ГОСТ 29227 и ГОСТ 29228.
Кислота серная по ГОСТ 4204, раствор концентрацией 5 М.
Натрий молибденовокислый (молибдат натрия) по ГОСТ 10931:
5 г молибденовокислого натрия (молибдата натрия) растворяют в 200 мл раствора серной кислоты концентрацией 5 М.
Гидразин сернокислый по ГОСТ 5841, 0,15 %-ный водный раствор (далее — восстановитель).
Калия гидроокись (гидроксид калия) по ГОСТ 24363, раствор концентрацией 3 М.
Смесь для сплавления по 4.11.1.
Кислота соляная по ГОСТ 3118, раствор 1:5.
Калий фосфорнокислый (фосфат калия) однозамещенный по ГОСТ 4198, дважды перекристаллизованный из горячего водного раствора и высушенный до постоянной массы при температуре 11 °С. Хранят в эксикаторе над слоем серной кислоты или в стеклянной таре с притертой крышкой.
Индикатор фенолфталеин.
4.14.2 Порядок подготовки к проведению анализа
Приготовление стандартного раствора
Навеску однозамещенного фосфорнокислого калия (фосфата калия) массой 0,1917 г растворяют в 100—150 мл воды, переносят в мерную колбу вместимостью 1 л, разбавляют до метки водой и перемешивают. Затем 100 мл приготовленного раствора переносят в мерную колбу вместимостью 500 мл и доводят водой до 500 мл. Массовая концентрация оксида фосфора в стандартном растворе — 0,02 мг/мл.
Построение градуировочного графика
В четыре мерные колбы вместимостью по 50 мл вводят соответственно 1; 2; 3; 4 мл стандартного раствора, что соответствует 0,02; 0,04; 0,06; 0,08 мг оксида фосфора, добавляют в каждую колбу 25— 30 мл воды, 5 мл раствора молибденовокислого натрия, 2 мл сернокислого гидразина и доливают до метки водой. Растворы перемешивают и погружают на 10 мин в кипящую водяную баню, после чего охлаждают до комнатной температуры и полученные градуировочные растворы фотометрируют относительно дистиллированной воды на фотоэлектроколориметре с красным светофильтром с максимальным светопропусканием при длине волны 656—680 нм или на спектрофотометре при длине волны 830 нм, используя кювету с толщиной слоя, поглощающего свет, 20 мм.
По полученным результатам определений оптической плотности и известной концентрации оксида фосфора в фотометрируемых растворах строят градуировочный график.
4.14.3 Порядок проведения анализа
Пипеткой отбирают 10—25 мл анализируемого раствора, переносят в мерную колбу вместимостью 50 мл. Предварительно 10—25 мм этого же раствора отбирают в стакан или колбу и нейтрализуют раствором гидроксида калия по фенолфталеину. Пошедшее на нейтрализацию количество щелочи прибавляют в анализируемый раствор, затем добавляют 5 мл раствора молибденовокислого натрия (молибдат натрия), 2 мл раствора сернокислого гидразина, доливают до метки водой, перемешивают и через 10 мин выдерживания в кипящей водяной бане охлаждают. Фотометрируют на фотоэлектроколориметре с красным светофильтром с максимальным светопропусканием при длине волны 656—680 нм или на спектрофотометре при длине волны 830 нм, используя кювету с толщиной слоя, поглощающего свет, 20 мм.
4.14.4 Обработка результатов анализа
Массовую долю оксида фосфора Р2О5 в исследуемом материале, %, определяют по формуле
![]() (38)
(38)
где V — объем окрашенного исследуемого раствора, мл;
Vо — общий объем исследуемого раствора, мл;
т — масса навески, г;
Va — объем аликвотной части исследуемого раствора, взятой для приготовления окрашенного раствора, мл;
Сx — концентрация оксида фосфора в окрашенном исследуемом растворе, мг/мл, найденная по градуировочному графику или методом сравнения по формуле
![]() (39)
(39)
где Сс — концентрация раствора сравнения, мг/мл;
Дх — оптическая плотность раствора сравнения;
Дс — оптическая плотность окрашенного исследуемого раствора. Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 16.
Таблица 16 В процентах
Массовая доля оксида фосфора | Абсолютное допустимое расхождение |
До 0,5 включ. Св. 0,5 » 1,0 » » 1,0 » 3,0 » | 0,04 0,10 0,25 |
4.15 Определение суммы оксида и гидроксида кальция (свободной извести или свободного оксида кальция)
4.15.1 Этанол-глицератный метод
Метод основан на экстрагировании оксида и гидроксида кальция из свежерастертого порошка анализируемой пробы этанол-глицериновым раствором (далее — растворителем) с последующим титрованием образовавшегося глицерата кальция спиртовым раствором безводной бензойной кислоты в присутствии индикатора фенолфталеина.
4.15.1.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104.
Печь муфельная.
Шкаф сушильный.
Холодильник Либиха.
Холодильник обратный.
Бюретка по ГОСТ 29252.
Круглодонные колбы вместимостью 150, 200 мл, 2, 3 л по ГОСТ 25336.
Спиртометр.
Кальция оксид по ГОСТ 8677.
Кальций углекислый (карбонат кальция) по ГОСТ 4530.
Эксикатор по ГОСТ 25336.
Баня водяная.
Баня песчаная.
Колбы конические вместимостью 150, 200, 300, 350, 500 мл по ГОСТ 25336.
Стакан термостойкий вместимостью 300 мл по ГОСТ 25336.
Тигель платиновый по ГОСТ 6563.
Свежеобожженный оксид кальция: оксид кальция или углекислый кальций (карбонат кальция) прокаливают в муфельной печи не менее 4 ч при температуре не менее 950 °С; обожженный продукт охлаждают в эксикаторе и немедленно используют для обезвоживания спирта.
Спирт этиловый ректификованный технический по ГОСТ 18300.
Натрия гидроокись (гидроксид натрия) по ГОСТ 4328, спиртовой раствор концентрацией 0,1 моль/л (0,1 М).
Глицерин по ГОСТ 6259.
Кислота бензойная по ГОСТ 10521, спиртовой раствор концентрацией 0,1 моль/л: 12,3 г обезвоженной бензойной кислоты растворяют в 1 л абсолютного этилового спирта.
Бария хлорид по ГОСТ 4108.
Индикатор фенолфталеин.
Трубка хлоркальциевая.
Известь натронная.
Стеклянные бусинки или отмытый и затем прокаленный кварцевый песок.
4.15.1.2 Порядок подготовки к проведению анализа
Обезвоживание реагентов
Абсолютный спирт готовят следующим образом: в круглодонную колбу вместимостью 2—3 л насыпают до половины свежеобожженный оксид кальция, заливают на 2/3 колбы этиловым спиртом, плотно закрывают колбу резиновой пробкой с хлоркальциевой трубкой.
Спирт настаивают над оксидом кальция в течение 2—3 сут. По истечении этого времени колбу соединяют герметично с обратным холодильником, ставят на кипящую водяную баню и кипятят содержимое в течение 5—6 ч. Водяную баню нагревают электроплиткой с закрытой спиралью во избежание воспламенения паров спирта.
По окончании кипячения снимают обратный холодильник и закрывают колбу резиновой пробкой, в центральное отверстие которой вставляют изогнутую трубку. Присоединяют колбу к холодильнику Либиха, на выходе которого должен быть алонж, соединенный с сухой колбой вместимостью 1 л через резиновую пробку с двумя отверстиями. Во второе отверстие вставляют хлоркальциевую трубку. Все соединения должны быть строго герметичными во избежание потерь спирта в атмосферу.
Первыми 15—20 мл перегнанного спирта ополаскивают приемник; последнюю порцию отгона, около 20 мл, не применяют для приготовления титрованных растворов.
Полученный безводный спирт охлаждают до температуры 20 °С и проверяют его концентрацию спиртометром. Если спирта окажется менее 99,8 %, то его вторично настаивают над новой порцией свежеобожженного оксида кальция и отгоняют.
Полученный безводный спирт хранят в герметично закрытых бутылях.
Безводный глицерин готовят следующим образом: в термостойкий стакан вместимостью 300 мл наливают 250—300 мл глицерина и нагревают в течение 3 ч при температуре 160—170 °С. Температуру проверяют термометром, опущенным в глицерин.
Обезвоженный и охлажденный глицерин переливают в сухую колбу вместимостью 250—300 мл и герметично закрывают пробкой.
Безводную бензойную кислоту получают высушиванием до постоянной массы в течение суток в эксикаторе над серной кислотой.
Безводный хлорид бария готовят высушиванием в сушильном шкафу при температуре 130 °С.
Приготовление растворителей
Этанол-глицериновый растворитель готовят следующим образом:
в 200 мл безводного глицерина, нагретого в конической колбе до температуры 100—125 °С, растворяют 15 г безводного хлорида бария. Раствор охлаждают и прибавляют 1 л безводного спирта и 0,1 г индикатора фенолфталеина. Полученный растворитель нейтрализуют, прибавляя каплями спиртовые растворы гидроксида натрия и бензойной кислоты, добиваясь бледно-розовой окраски.
Определение титра раствора бензойной кислоты по оксиду кальция
Навеску оксида кальция или углекислого кальция (карбоната кальция) массой 0,2 г прокаливают до постоянной массы в течение 2—3 ч при температуре 950—1000 °С в платиновом тигле. Полученный оксид растирают и вновь прокаливают не менее 30 мин при той же температуре, затем охлаждают в эксикаторе над натронной известью.
В три сухие круглодонные или конические колбы вместимостью 150 мл помещают стеклянные бусинки, наливают 30—40 мл растворителя, быстро вносят отвешенную в закрытом бюксе навеску свежеприготовленного оксида кальция массой 0,03—0,04 г, энергично взбалтывают и герметично присоединяют к обратному холодильнику.
Раствор нагревают до кипения на песчаной бане и кипятят до появления интенсивной розовой окраски. Затем колбу отсоединяют от холодильника и тотчас же титруют ее содержимое раствором бензойной кислоты до исчезновения окраски. Снова присоединяют колбу к обратному холодильнику и кипятят раствор. Кипячение и титрование чередуют до исчезновения окраски при последующем кипячении в течение 20—30 мин.
Титр раствора бензойной кислоты ТCaO ,г/мл, определяют по формуле
![]() , (40)
, (40)
где т — масса навески оксида кальция, г;
V — объем раствора бензойной кислоты, пошедший на титрование, мл.
За значение титра принимают среднеарифметическое не менее трех титрований.
4.15.1.3 Порядок проведения анализа
В зависимости от предполагаемой массовой доли свободного оксида кальция навеску свежерастертой пробы массой 0,1—1,0 г помещают в коническую или круглодонную колбу вместимостью 150— 200 мл и прибавляют 30—40 мл растворителя. Содержимое колбы взбалтывают. Колбу присоединяют к обратному холодильнику и кипятят 20—30 мин на песчаной бане. При появлении розовой окраски раствора колбу отсоединяют от холодильника и тотчас же титруют горячий раствор раствором бензойной кислоты. Кипячение и титрование чередуют до исчезновения окраски при последующем кипячении в течение 20—30 мин, после чего титрование считают законченным.
4.15.1.4 Обработка результатов анализа
Массовую долю оксида и гидроксида кальция (свободной извести) СаОсв, %, определяют по формуле
![]() , (41)
, (41)
где V — объем раствора бензойной кислоты, пошедший на титрование анализируемого раствора, мл;
TСаO— титр раствора бензойной кислоты по СаО, г/мл;
т — масса навески, г.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 17.
Таблица 17 В процентах
Массовая доля свободного оксида кальция | Абсолютное допустимое расхождение |
До 1,0 включ. Св. 1,0 » 2,0 » » 2,0 » 10,0 » » 10,0 » 25,0 » | 0,06 0,10 0,40 0,69 |
4.15.2 Этиленгликолевый метод
Метод основан на экстрагировании суммы оксида и гидроксида кальция (свободной извести) из свежерастертой анализируемой пробы этиленгликолем с последующим титрованием образовавшегося этиленгликолята кальция соляной кислотой с комбинированным индикатором (фенолфталеин—(—нафтолфталеин), резко изменяющим окраску в узком интервале рН. Определение проводят, применяя навеску, находящуюся во влажно-сухом состоянии.
4.15.2.1 Средства контроля и вспомогательное оборудование
Весы аналитические по ГОСТ 24104.
Шкаф сушильный.
Бюксы по ГОСТ 23932.
Насос водоструйный по ГОСТ 25336.
Воронка с фильтром Шотта № 2 по ГОСТ 25336.
Баня водяная.
Колбы конические вместимостью 250 мл по ГОСТ 25336.
Бюретка по ГОСТ 29252.
Соляная кислота по ГОСТ 3118, титрованный 0,1 М раствор.
Этиленгликоль по ГОСТ 10164.
Индикатор: 0,1 г фенолфталеина и 0,15 г (-нафтолфталеина растворяют в 100 мл этиленгликоля.
Стеклянные бусинки или отмытый и затем прокаленный кварцевый песок.
Эксикатор по ГОСТ 25336.
Кальций хлористый (хлорид кальция) по ГОСТ 450, прокаленный при температуре 700—800 °С, для заполнения эксикатора.
4.15.2.2 Порядок проведения анализа
Пробу измельчают до состояния пудры (при контрольном просеивании проба должна полностью пройти через сито № 008 по ГОСТ 6613). Отбирают две навески: одну массой 1,0 г для определения влаги по 4.2; другую — массой 0,5—1,0 г — для определения содержания свободной извести. Эту навеску помещают в коническую колбу вместимостью 250 мл и заливают 40—50 мл этиленгликоля. Для лучшего перемешивания в колбу помещают 4—5 стеклянных бусинок или зерна кварца. Колбу закрывают резиновой пробкой, ставят на 30 мин в водяную баню, температура которой должна быть не более 70 °С. Содержимое колбы непрерывно помешивают. Затем раствор фильтруют через воронку с фильтром Шотта под разряжением.
Фильтр после фильтрования 3 раза промывают этиленгликолем. В фильтрат добавляют 2—3 капли индикатора и титруют соляной кислотой до обесцвечивания раствора.
4.15.2.3 Обработка результатов анализа
Массовую долю суммы оксида и гидроксида кальция (свободной извести) СаОсв, % , определяют по формуле
![]() , (42)
, (42)
где V — объем соляной кислоты, израсходованный на титрование, мл;
ТСaO— титр раствора соляной кислоты по СаО, г/мл;
т — масса воздушно-сухой навески, г;
W — содержание влаги, %, определенное по 4.2.
Абсолютное допустимое расхождение результатов параллельных определений не должно превышать значений, указанных в таблице 17.
ПРИЛОЖЕНИЕ А
(информационное)
Нормативные ссылки
В настоящем стандарте использованы ссылки на следующие нормативные документы:
ГОСТ 8.315—97 ГСИ. Стандартные образцы. Основные положения, порядок разработки, аттестации, утверждения, регистрации и применения
ГОСТ 8.326—89 ГСИ. Метрологическая аттестация средств измерения
ГОСТ 8.513—84 ГСИ. Поверка средств измерения. Организация и порядок проведения
ГОСТ 8.532—85 ГСИ. Стандартные образцы состава веществ и материалов. Порядок межлабораторной аттестации
ГОСТ 12.1.004—91 ССБТ. Пожарная безопасность. Общие требования
ГОСТ 12.1.005—88 ССБТ. Общие санитарно-гигиенические требования к воздуху в рабочей зоне
ГОСТ 12.1.007—76 ССБТ. Вредные вещества. Классификация и общие требования безопасности
ГОСТ 12.1.010—76 ССБТ. Взрывобезопасность. Общие требования
ГОСТ 12.1.019—79 ССБТ. Электробезопасность. Общие требования и номенклатура видов защиты
ГОСТ 12.4.011—89 ССБТ. Средства защиты работающих. Общие требования и классификация
ГОСТ 12.4.028—76 ССБТ. Респираторы ШБ-1 «Лепесток». Технические условия
ГОСТ 12.4.103—83 ССБТ. Одежда специальная защитная, средства индивидуальной защиты ног и рук. Классификация .
ГОСТ 83—79 Натрий углекислый. Технические условия
ГОСТ 199—78 Натрий уксуснокислый 3-водный. Технические условия
ГОСТ 450—77 Кальций хлористый технический. Технические условия
ГОСТ 1277—75 Серебро азотнокислое. Технические условия
ГОСТ 1770—74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Технические условия
ГОСТ 3118—77 Кислота соляная. Технические условия
ГОСТ 3760—79 Аммиак водный. Технические условия
ГОСТ 3762—78 Аммоний углекислый. Технические условия
ГОСТ 3773—72 Аммоний хлористый. Технические условия
ГОСТ 4108—72 Барий хлорид 2-водный. Технические условия
ГОСТ 4139—75 Калий роданистый. Технические условия
ГОСТ 4145—74 Калий сернокислый. Технические условия
ГОСТ 4147—74 Железо (III) хлорид 6-водный. Технические условия
ГОСТ 4159—79 Йод. Технические условия
ГОСТ 4166—76 Натрий сернокислый. Технические условия
ГОСТ 4198—75 Калий фосфорнокислый однозамещенный. Технические условия
ГОСТ 4199—76 Натрий тетраборнокислый 10-водный. Технические условия.
ГОСТ 4204—77 Кислота серная. Технические условия.
ГОСТ 4217—77 Калий азотнокислый. Технические условия
ГОСТ 4220—75 Калий двухромовокислый. Технические условия.
ГОСТ 4221—76 Калий углекислый. Технические условия
ГОСТ 4232—74 Калий йодистый. Технические условия
ГОСТ 4233—77 Натрий хлористый. Технические условия
ГОСТ 4328—77 Натрия гидроокись. Технические условия
ГОСТ 4459—75 Калий хромовокислый. Технические условия
ГОСТ 4461—77 Кислота азотная. Технические условия
ГОСТ 4478—78 Кислота сульфосалициловая 2-водная. Технические условия
ГОСТ 4523—77 Магний сернокислый 7-водный. Технические условия
ГОСТ 4530—76 Кальций углекислый. Технические условия
ГОСТ 5456—79 Гидроксиламина гидрохлорид. Технические условия
ГОСТ 5833—75 Сахароза. Технические условия
ГОСТ 5841—74 Гидразин сернокислый
ГОСТ 6259—75 Глицерин. Технические условия
ГОСТ 6552—80 Кислота ортофосфорная. Технические условия
ГОСТ 6563—75 Изделия технические из благородных металлов и сплавов. Технические условия
ГОСТ 6613—86 Сетки проволочные тканые с квадратными ячейками. Технические условия
ГОСТ 6709—72 Вода дистиллированная. Технические условия
ГОСТ 8269.0—97 Щебень и гравий из плотных горных пород и отходов промышленного производства для строительных работ. Методы физико-механических испытаний
ГОСТ 8677—76 Кальция оксид. Технические условия
ГОСТ 9147—80 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 10163—76 Крахмал растворимый. Технические условия
ГОСТ 10164—75 Этиленгликоль. Технические условия
ГОСТ 10484—78 Кислота фтористоводородная. Технические условия
ГОСТ 10521—78 Кислота бензойная. Технические условия
ГОСТ 10652—73 Соль динатриевая этилендиамин — N, N, N1, N1 — тетрауксусной кислоты 2-водная (трилон Б)
ГОСТ 10929—76 Водорода пероксид. Технические условия
ГОСТ 10931—74 Натрий молибденовокислый 2-водный. Технические условия
ГОСТ 11069—74 Алюминий первичный. Марки
ГОСТ 18270—72 Кислота уксусная особой чистоты. Технические условия
ГОСТ 18300—87 Спирт этиловый ректификованный технический. Технические условия
ГОСТ 19908—90 Тигли, чаши, стаканы, колбы, воронки, пробирки и наконечники из прозрачного кварцевого песка. Общие технические условия
ГОСТ 20478—75 Аммоний надсернокислый. Технические условия
ГОСТ 20490—75 Калий марганцовокислый. Технические условия
ГОСТ 22867—77 Аммоний азотнокислый. Технические условия
ГОСТ 23932—90 Посуда и оборудование лабораторные стеклянные. Общие технические условия
ГОСТ 24104—88 Весы лабораторные общего назначения и образцовые. Общие технические условия
ГОСТ 24363—80 Калия гидроокись. Технические условия
ГОСТ 24555—81 Система государственных испытаний продукции. Порядок аттестации испытательного оборудования. Основные положения
ГОСТ 25336—82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 27067—86 Аммоний роданистый. Технические условия
ГОСТ 27068—86 Натрий серноватистокислый (натрия тиосульфат) 5-водный. Технические условия
ГОСТ 27654—88 Костюмы женские для защиты от кислот. Технические условия
ГОСТ 29058—91 Костюмы женские для защиты от нетоксичной пыли. Технические условия
ГОСТ 29227—91 Посуда лабораторная стеклянная. Пипетки градуированные. Часть 1. Общие требования
ГОСТ 29228—91 Посуда лабораторная стеклянная. Пипетки градуированные. Часть 2. Пипетки градуированные без установленного времени ожидания
ГОСТ 29230—91 Посуда лабораторная стеклянная. Пипетки градуированные. Часть 4. Пипетки выдувные
ГОСТ 29251—91 Посуда лабораторная стеклянная. Бюретки. Часть 1. Общие требования
ГОСТ 29252—91 Посуда лабораторная стеклянная. Бюретки. Часть 2. Общие требования. Бюретки без времени ожидания
УДК [б9+691.224:54](084.74) ОКС 91.100.20 Ж19 ОКСТУ 5709
Ключевые слова: щебень, гравий, горные породы, отходы промышленного производства, химический анализ, проба, навеска
СОДЕРЖАНИЕ
1 Область применения
2 Нормативные ссылки
3 Термины и определения
4 Методы химического анализа
4.1 Общие положения
4.2 Определение влаги
4.3 Определение потери массы при прокаливании
4.4 Определение диоксида кремния
4.5 Определение оксидов железа и алюминия
4.6 Определение оксидов кальция и магния
4.7 Определение сульфатной и сульфидной серы
4.8 Определение оксидов калия и натрия
4.9 Определение оксида железа двухвалентного
4.10 Определение общего содержания хлоридов и легкорастворимых хлоридов
4.11 Определение оксида марганца
4.12 Определение диоксида титана
4.13 Определение оксида хрома
4.14 Определение оксида фосфора
4.15 Определение суммы оксида и гидроксида кальция (свободной извести или свободного оксида кальция)
Приложение А Нормативные ссылки